

Role of Alpha-Fetoprotein (AFP) in Diagnosing Childhood Cancers and Genetic-Related Chronic Diseases
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. AFP—Diagnostic Difficulties in Pediatrics
2.1. Postpartum AFP Concentrations
2.2. Methods of Determination and Establishment of AFP Reference Values
2.3. Comparing the Results of AFP Concentrations
2.4. Size of Study Groups
3. Liver
3.1. Ataxia Telangiectasia (AT)
3.2. Primrose Syndrome
3.3. Type I Tyrosinemia
3.4. Progressive Familial Intrahepatic Cholestasis—PFIC2
3.5. Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency—NICCD
3.6. Transaldolase Deficiency—TALDO
3.7. Hepatitis B (HBV) in Children
4. Hematopoietic System
Fanconi Anemia
5. Endocrine System
Hypothyroidism
6. Cancers
6.1. Hepatoblastoma
6.2. Hepatocellular Carcinoma
6.3. Germ Cell Tumors
6.4. Intracranial Germ Cell Tumors (IC-GCTs)
6.5. Malignant Saccrococygeal Germ Cell Tumor
6.6. Special Histopathological Cases of GCT Connected with High AFP Levels
6.6.1. Yolk Sac Tumor (YST = Endodermal Sinus Tumor)
6.6.2. Embryonal Carcinoma (EC)
7. Ovarian Sertoli-Leydig Cell Tumor (SLCT)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFP | alpha-fetoprotein |
| AT | ataxia-telangiectasia |
| AOA | ataxia with oculomotor apraxia |
| BRCA2 | BReast CAncer gene 2 |
| CR | complete remission |
| CSF | cerebrospinal fluid |
| EC | embryonal carcinoma |
| EGGCT | extragonadal germ cell tumors |
| EFS | event-free survival |
| FA | Fanconi anemia |
| FHSC | fetal hepatic stem/progenitor cells |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HB | hepatoblastoma |
| HOC | hepatic oval cell |
| HSPC | hematopoietic stem/progenitor cells |
| GCTs | germ cell tumors |
| IC-GCT | intracranial germ cell tumor |
| LF/MCT | medium-chain triglyceride |
| NICCD | neonatal intrahepatic cholestasis caused by citrin deficiency |
| NTBC | 2-(2-nitro-4-3 trifluoro-methylbenzoyl)-1,3-cyclohexanedione |
| MGCT | mixed germ cell tumor |
| miRNA | micro ribonucleic acid |
| OS | overall survival |
| OYST | ovarian yolk sac tumor |
| PGC | primordial germ cell |
| PFIC-2 | progressive familial intrahepatic cholestasis type 2 |
| PUFA | polyunsaturated fatty acid |
| RIA | radioimmunoassay |
| RFS | relapse-free survival |
| SCT | sacrococcygeal teratoma |
| SLCT | Sertoli–Leydig cell tumor |
| T3 | triiodothyronine |
| T4 | thyroxine |
| TALDO | transaldolase deficiency |
| VATER/VACTERL | VATER/VACTERL association |
| YST | yolk sac tumor |
References
- Mizejewski, G.J. Levels of alpha-fetoprotein during pregnancy and early infancy in normal and disease states. Obstet. Gynecol. Surv. 2003, 58, 804–826. [Google Scholar] [CrossRef]
- Blohm, M.E.; Vesterling-Hörner, D.; Calaminus, G.; Göbel, U. Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr. Hematol. Oncol. 1998, 15, 135–142. [Google Scholar] [CrossRef]
- Ferraro, S.; Panzeri, A.; Braga, F.; Panteghini, M. Serum α-fetoprotein in pediatric oncology: Not a children’s tale. Clin. Chem. Lab. Med. 2019, 57, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Dall’Igna, P.; Brugieres, L.; Christin, A.S.; Maibach, R.; Casanova, M.; Alaggio, R.; de Goyet, J.V.; Zsiros, J.; Morland, B.; Czauderna, P.; et al. Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatr. Blood Cancer 2018, 65, e26791. [Google Scholar] [CrossRef]
- Frazier, A.L.; Hale, J.P.; Rodriguez-Galindo, C.; Dang, H.; Olson, T.; Murray, M.J.; Amatruda, J.F.; Thornton, C.; Arul, G.S.; Billmire, D.; et al. Revised risk classification for pediatric extracranial germ cell tumors based on 25 years of clinical trial data from the United Kingdom and United States. J. Clin. Oncol. 2015, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Cho, M.J.; Kim, D.Y.; Kim, S.C. Half-life of alpha-fetoprotein in neonatal sacrococcygeal teratoma. J. Pediatr. Surg. 2018, 53, 2470–2474. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.A.; Rothblum-Oviatt, C.C.; Wright, J.; Schlechter, H.; Lefton-Greif, M.A.; Natale, V.A.; Crawford, T.O.; Lederman, H.M. Multidisciplinary Management of Ataxia Telangiectasia: Current Perspectives. J. Multidiscip. Healthc. 2021, 14, 1637–1644. [Google Scholar] [CrossRef]
- Schieving, J.H.; de Vries, M.; van Vugt, J.M.; Weemaes, C.; van Deuren, M.; Nicolai, J.; Wevers, R.A.; Willemsen, M.A. Alpha-fetoprotein, a fascinating protein and biomarker in neurology. Eur. J. Paediatr. Neurol. 2014, 18, 243–248. [Google Scholar] [CrossRef]
- Renaud, M.; Tranchant, C.; Koenig, M.; Anheim, M. Autosomal Recessive Cerebellar Ataxias With Elevated Alpha-Fetoprotein: Uncommon Diseases, Common Biomarker. Mov. Disord. 2020, 35, 2139–2149. [Google Scholar] [CrossRef]
- Devaney, R.; Pasalodos, S.; Suri, M.; Bush, A.; Bhatt, J.M. Ataxia telangiectasia: Presentation and diagnostic delay. Arch. Dis. Child. 2017, 102, 328–330. [Google Scholar] [CrossRef]
- Stray-Pedersen, A.; Borresen-Dale, A.L.; Paus, E.; Lindman, C.R.; Burgers, T.; Abrahamsen, T.G. Alpha fetoprotein is increasing with age in ataxia-telangiectasia. Eur. J. Paediatr. Neurol. 2007, 11, 375–380. [Google Scholar] [CrossRef]
- Lynch, D.R.; McCormick, A.; Schadt, K.; Kichula, E. Pediatric Ataxia: Focus on Chronic Disorders. Semin. Pediatr. Neurol. 2018, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Melis, D.; Carvalho, D.; Barbaro-Dieber, T.; Espay, A.J.; Gambello, M.J.; Gener, B.; Gerkes, E.; Hitzert, M.M.; Hove, H.B.; Jansen, S.; et al. Primrose syndrome: Characterization of the phenotype in 42 patients. Clin. Genet. 2020, 97, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.J.; Netzel, B.C.; Singh, R.H.; Pino, G.B.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Rinaldo, P.; Tortorelli, S.; Smith, W.E.; et al. Laboratory monitoring of patients with hereditary tyrosinemia type I. Mol. Genet. Metab. 2020, 130, 247–254. [Google Scholar] [CrossRef] [PubMed]
- van Ginkel, W.G.; Pennings, J.P.; van Spronsen, F.J. Liver Cancer in Tyrosinemia Type 1. Adv. Exp. Med. Biol. 2017, 959, 101–109. [Google Scholar] [CrossRef]
- Bhushan, S.; Noble, C.; Balouch, F.; Lewindon, P.; Lampe, G.; Hodgkinson, P.; McGill, J.; Ee, L. Hepatocellular carcinoma requiring liver transplantation in hereditary tyrosinemia type 1 despite nitisinone therapy and α1-fetoprotein normalization. Pediatr. Transplant. 2022, 26, e14334. [Google Scholar] [CrossRef]
- Khanna, R.; Verma, S.K. Pediatric hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 3980–3999. [Google Scholar] [CrossRef]
- Koelink, C.J.; van Hasselt, P.; van der Ploeg, A.; van den Heuvel-Eibrink, M.M.; Wijburg, F.A.; Bijleveld, C.M.; van Spronsen, F.J. Tyrosinemia type I treated by NTBC: How does AFP predict liver cancer? Mol. Genet. Metab. 2006, 89, 310–315. [Google Scholar] [CrossRef]
- El-Karaksy, H.; Abdullatif, H.M.; Ghobrial, C.M.; Mogahed, E.A.; Yasin, N.A.; Talal, N.; Rashed, M. Clinical experience with hepatorenal tyrosinemia from a single Egyptian center. PLoS ONE 2022, 17, e0268017. [Google Scholar] [CrossRef]
- Fuenzalida, K.; Leal-Witt, M.J.; Guerrero, P.; Hamilton, V.; Salazar, M.F.; Peñaloza, F.; Arias, C.; Cornejo, V. NTBC Treatment Monitoring in Chilean Patients with Tyrosinemia Type 1 and Its Association with Biochemical Parameters and Liver Biomarkers. J. Clin. Med. 2021, 10, 5832. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, S.; Salo, M.K.; Kuusela, P.; Holmberg, C.; Simell, O.; Heikinheimo, M. Serum levels of oncofetal markers CA 125, CA 19-9, and alpha-fetoprotein in children with hereditary tyrosinemia type I. Pediatr. Res. 1994, 35, 205–208. [Google Scholar] [CrossRef]
- Srivastava, A. Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36. [Google Scholar] [CrossRef]
- Vinayagamoorthy, V.; Srivastava, A.; Sarma, M.S. Newer variants of progressive familial intrahepatic cholestasis. World J. Hepatol. 2021, 13, 2024–2038. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Song, Y.Z. Citrin Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Lipiński, P.; Jurkiewicz, D.; Ciara, E.; Płoski, R.; Więcek, S.; Bogdańska, A.; Stradomska, T.; Socha, P.; Rokicki, D.; Tylki-Szymańska, A.; et al. Neonatal cholestasis due to citrin deficiency: Diagnostic pitfalls. Acta Biochim. Pol. 2020, 67, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, K. Metabolic basis and treatment of citrin deficiency. J. Inherit. Metab. Dis. 2021, 44, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Valayannopoulos, V.; Altassan, R.; Chung, W.K.; Heijboer, A.C.; Keng, W.T.; Lapatto, R.; McClean, P.; Mulder, M.F.; Tylki-Szymańska, A.; et al. Clinical, biochemical, and molecular overview of transaldolase deficiency and evaluation of the endocrine function: Update of 34 patients. J. Inherit. Metab. Dis. 2019, 42, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Rodan, L.H.; Berry, G.T. N-Acetylcysteine Therapy in an Infant with Transaldolase Deficiency Is Well Tolerated and Associated with Normalization of Alpha Fetoprotein Levels. JIMD Rep. 2017, 31, 73–77. [Google Scholar] [CrossRef]
- Lipiński, P.; Stradomska, T.; Tylki-Szymańska, A. Transaldolase deficiency-clinical outcome, pathogenesis, diagnostic process. Dev. Period. Med. 2018, 22, 187–196. (In Polish) [Google Scholar] [CrossRef]
- Grammatikopoulos, T.; Hadzic, N.; Foskett, P.; Strautnieks, S.; Samyn, M.; Vara, R.; Dhawan, A.; Hertecant, J.; Al Jasmi, F.; Rahman, O.; et al. Liver Disease and Risk of Hepatocellular Carcinoma in Children With Mutations in TALDO1. Hepatol. Commun. 2022, 6, 473–479. [Google Scholar] [CrossRef]
- Zong, X.; Yang, J.X.; Zhang, Y. Persistently elevated alpha-fetoprotein associated with chronic hepatitis B during chemotherapy for malignant ovarian germ cell tumors: A case series and a review of the literature. J. Ovarian Res. 2019, 12, 124. [Google Scholar] [CrossRef]
- Kim, C.Y.; Kim, B.R.; Lee, S.S.; Jeon, D.H.; Lee, C.M.; Kim, W.S.; Cho, H.C.; Kim, J.J.; Lee, J.M.; Kim, H.J.; et al. Clinical features of hepatitis B and C virus infections, with high α-fetoprotein levels but not hepatocellular carcinoma. Medicine 2017, 96, e5844. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, N. Hepatitis B in children: Complexities in management. Pediatr. Transplant. 2005, 9, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.P.Y.; Soh, S.Y.; Cheng, F.W.C.; Pang, H.H.; Luk, C.W.; Li, C.H.; Ho, K.K.H.; Chan, E.K.W.; Chan, A.C.Y.; Chung, P.H.Y.; et al. Hepatitis B Virus Seropositivity Is a Poor Prognostic Factor of Pediatric Hepatocellular Carcinoma: A Population-Based Study in Hong Kong and Singapore. Front. Oncol. 2020, 10, 570479. [Google Scholar] [CrossRef] [PubMed]
- Aslan, D.; Karabacak, R.O.; Aslan, O.D. Maternal serum alpha-fetoprotein levels are normal in Fanconi anemia: Can it be a lack of postnatal inhibition of AFP gene resulting in the elevation? Pediatr. Blood Cancer 2017, 64, e26297. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N. Serum alpha fetoprotein levels in Fanconi anaemia. Br. J. Haematol. 2019, 184, 1074–1076. [Google Scholar] [CrossRef]
- Cassinat, B.; Guardiola, P.; Chevret, S.; Schlageter, M.H.; Toubert, M.E.; Rain, J.D.; Gluckman, E. Constitutive elevation of serum alpha-fetoprotein in Fanconi anemia. Blood 2000, 96, 859–863. [Google Scholar] [CrossRef]
- Salem, B.; Mitchell, R.; DeFor, T.E.; Tryon, R.; Wagner, J.E.; MacMillan, M.L. Elevations in serum alpha fetoprotein levels in patients with Fanconi anaemia. Br. J. Haematol. 2019, 184, 1032–1035. [Google Scholar] [CrossRef]
- Lakhi, N.A.; Mizejewski, G.J. Alpha-fetoprotein and Fanconi Anemia: Relevance to DNA Repair and Breast Cancer Susceptibility. Fetal Pediatr. Pathol. 2017, 36, 49–61. [Google Scholar] [CrossRef]
- Anteby, E.; Shpan, P.; Dushnik, M.; Zvang, A.; Zer, T.; Ben-Neriah, Z.; Yagel, S. The regulatory role of tri-iodothyronine on the production of alpha-fetoprotein and albumin by mouse fetal liver cells. Hum. Reprod. 1993, 8, 1576–1578. [Google Scholar] [CrossRef]
- László, V.; Dezso, K.; Baghy, K.; Papp, V.; Kovalszky, I.; Sáfrány, G.; Thorgeirsson, S.S.; Nagy, P.; Paku, S. Triiodothyronine accelerates differentiation of rat liver progenitor cells into hepatocytes. Histochem. Cell Biol. 2008, 130, 1005–1014. [Google Scholar] [CrossRef]
- Patni, N.; Cervantes, L.F.; Diaz, A. Elevated alpha-fetoprotein levels in Van Wyk-Grumbach syndrome: A case report and review of literature. J. Pediatr. Endocrinol. Metab. 2012, 25, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Lin-Su, K. Van Wyk-Grumbach syndrome in a female pediatric patient with trisomy 21: A case report. Int. J. Pediatr. Endocrinol. 2020, 2020, 2. [Google Scholar] [CrossRef]
- Baranowski, E.; Högler, W. An unusual presentation of acquired hypothyroidism: The Van Wyk-Grumbach syndrome. Eur. J. Endocrinol. 2012, 166, 537–542. [Google Scholar] [CrossRef]
- Thompson, P.A.; Chintagumpala, M. Renal and hepatic tumors in the neonatal period. Semin. Fetal Neonatal Med. 2012, 17, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.L. Tumors of the liver in children. Surg. Oncol. 2007, 16, 195–203. [Google Scholar] [CrossRef]
- Tanimura, M.; Matsui, I.; Abe, J.; Ikeda, H.; Kobayashi, N.; Ohira, M.; Yokoyama, M.; Kaneko, M. Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res. 1998, 58, 3032–3035. [Google Scholar] [PubMed]
- Isaacs, H., Jr. Fetal and neonatal hepatic tumors. J. Pediatr. Surg. 2007, 42, 1797–1803. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.D.; Maibach, R.; Aronson, D.C.; Rangaswami, A.; Häberle, B.; O’Neill, A.F.; Schmid, I.; Ansari, M.; Hishiki, T.; Ranganathan, S.; et al. Outcomes of Patients Treated for Hepatoblastoma with Low Alpha-Fetoprotein and/or Small Cell Undifferentiated Histology: A Report from the Children’s Hepatic Tumors International Collaboration (CHIC). Cancers 2023, 15, 467. [Google Scholar] [CrossRef]
- Li, F.; Zhang, W.; Hu, H.; Zhu, X.; Zhang, Y.; Huang, D. Factors influencing recurrence after complete remission in children with hepatoblastoma: A 14-year retrospective study in China. PLoS ONE 2021, 16, e0259503. [Google Scholar] [CrossRef]
- Sintusek, P.; Phewplung, T.; Sanpavat, A.; Poovorawan, Y. Liver tumors in children with chronic liver diseases. World J. Gastrointest. Oncol. 2021, 13, 1680–1695. [Google Scholar] [CrossRef]
- Short, S.S.; Kastenberg, Z.J.; Wei, G.; Bondoc, A.; Dasgupta, R.; Tiao, G.M.; Watters, E.; Heaton, T.E.; Lotakis, D.; La Quaglia, M.P.; et al. Histologic type predicts disparate outcomes in pediatric hepatocellular neoplasms: A Pediatric Surgical Oncology Research Collaborative study. Cancer 2022, 128, 2786–2795. [Google Scholar] [CrossRef] [PubMed]
- Weeda, V.B.; Aronson, D.C.; Verheij, J.; Lamers, W.H. Is hepatocellular carcinoma the same disease in children and adults? Comparison of histology, molecular background, and treatment in pediatric and adult patients. Pediatr. Blood Cancer 2019, 66, e27475. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.; Frazier, A.L.; Shaikh, F. Germ Cell Tumors in Adolescents and Young Adults. J. Oncol. Pract. 2019, 15, 433–441. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.K.; Heerema-McKenney, A.; Rouse, R.V. Extragonadal germ cell tumors: A review with emphasis on pathologic features, clinical prognostic variables, and differential diagnostic considerations. Adv. Anat. Pathol. 2007, 14, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, H., Jr. Perinatal (fetal and neonatal) germ cell tumors. J. Pediatr. Surg. 2004, 39, 1003–1013. [Google Scholar] [CrossRef]
- Ataikiru, U.O.; Iacob, E.R.; Miron, I.; Popoiu, C.M.; Boia, E.S. A 10-year retrospective single-center study of alpha-fetoprotein and beta-human chorionic gonadotropin in Romanian children with (para)gonadal tumors and cysts. J. Pediatr. Endocrinol. Metab. 2021, 35, 363–371. [Google Scholar] [CrossRef]
- Perkins, G.L.; Slater, E.D.; Sanders, G.K.; Prichard, J.G. Serum tumor markers. Am. Fam. Physician 2003, 68, 1075–1082. [Google Scholar]
- Ehrlich, Y.; Beck, S.D.; Foster, R.S.; Bihrle, R.; Einhorn, L.H. Serum tumor markers in testicular cancer. Urol. Oncol. 2013, 31, 17–23. [Google Scholar] [CrossRef]
- D’Angelo, P.; De Pasquale, M.D.; Barretta, F.; Affinita, M.C.; Conte, M.; Dall’Igna, P.; Di Cataldo, A.; Inserra, A.; Provenzi, M.; Quaglietta, L.; et al. Malignant sacrococcygeal germ cell tumors in childhood: The Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) experience. Pediatr. Blood Cancer 2021, 68, e28812. [Google Scholar] [CrossRef]
- Fresneau, B.; Orbach, D.; Faure-Conter, C.; Sudour-Bonnange, H.; Vérité, C.; Gandemer, V.; Pasquet, M.; Fasola, S.; Rome, A.; Raimbault, S.; et al. Is alpha-fetoprotein decline a prognostic factor of childhood non-seminomatous germ cell tumours? Results of the French TGM95 study. Eur. J. Cancer 2018, 95, 11–19. [Google Scholar] [CrossRef]
- Faure-Conter, C.; Orbach, D.; Sudour-Bonnange, H.; Verité, C.; Mansuy, L.; Rome, A.; Dumesnil, C.; Thebaud, E.; Renard, M.; Hameury, F.; et al. Extracranial germ cell tumours in children and adolescents: Results from the French TGM13 protocol. Pediatr. Blood Cancer 2023, 70, e30117. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.F.; Xia, C.; Krailo, M.D.; Shaikh, F.; Pashankar, F.D.; Billmire, D.F.; Olson, T.A.; Amatruda, J.F.; Villaluna, D.; Huang, L.; et al. α-Fetoprotein as a predictor of outcome for children with germ cell tumors: A report from the Malignant Germ Cell International Consortium. Cancer 2019, 125, 3649–3656. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.M.; Tabernero, J.M.; Paz-Ares, L.; García-Llano, J.L.; Mora, J.; Lianes, P.; Esteban, E.; Salazar, R.; López-López, J.J.; Cortés-Funes, H. Tumor markers at the time of recurrence in patients with germ cell tumors. Cancer 2000, 88, 162–168. [Google Scholar] [CrossRef]
- Keskin, S.; Ekenel, M.; Başaran, M.; Bavbek, S. Predictive value of marker half-life in relapsed and nonrelapsed nonseminomatous germ cell testicular tumor patients undergoing chemotherapy. Am. J. Clin. Oncol. 2012, 35, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, M.; Gawrychowska, A.; Stefanowicz, J. Diagnostic, Prognostic and Predictive Markers in Pediatric Germ Cell Tumors-Past, Present and Future. Diagnostics 2022, 12, 278. [Google Scholar] [CrossRef]
- Saliyeva, S.; Boranbayeva, R.; Konoplya, N.; Bulegenova, M.; Blau, O.; Belousov, V.; Granica, J.; Mukushkina, D.; Altynbayeva, G. Pediatric Extracranial Germ Cell Tumors: Expression of microRNA. J. Pediatr. Hematol. Oncol. 2023, 45, e174–e179. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Bell, E.; Raby, K.L.; Rijlaarsdam, M.A.; Gillis, A.J.; Looijenga, L.H.; Brown, H.; Destenaves, B.; Nicholson, J.C.; Coleman, N. A pipeline to quantify serum and cerebrospinal fluid microRNAs for diagnosis and detection of relapse in paediatric malignant germ-cell tumours. Br. J. Cancer 2016, 114, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, R.; DaSilva, N.S.; Cappellano, A.; Belessiotis, C.; Diez, B.; Gardner, S.; Allen, J.; Weinblatt, M.; Gottardo, N.; Dhall, G.; et al. Relapse and outcome patterns of patients with central nervous system mixed malignant germ cell tumors treated without irradiation: Findings from the third international central nervous system (CNS) germ cell tumor (GCT) study. Pediatr. Blood Cancer 2015, 62, 1920–1924. [Google Scholar] [CrossRef]
- PDQ Pediatric Treatment Editorial Board. Childhood Central Nervous System Germ Cell Tumors Treatment (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Frappaz, D.; Dhall, G.; Murray, M.J.; Goldman, S.; Faure Conter, C.; Allen, J.; Kortmann, R.D.; Haas-Kogen, D.; Morana, G.; Finlay, J.; et al. EANO, SNO and Euracan consensus review on the current management and future development of intracranial germ cell tumors in adolescents and young adults. Neuro Oncol. 2022, 24, 516–527. [Google Scholar] [CrossRef]
- Calaminus, G.; Frappaz, D.; Kortmann, R.D.; Krefeld, B.; Saran, F.; Pietsch, T.; Vasiljevic, A.; Garre, M.L.; Ricardi, U.; Mann, J.R.; et al. Outcome of patients with intracranial non-germinomatous germ cell tumors-lessons from the SIOP-CNS-GCT-96 trial. Neuro Oncol. 2017, 19, 1661–1672. [Google Scholar] [CrossRef]
- Sathitsamitphong, L.; Monsereenusorn, C.; Techavichit, P.; Shotelersuk, K.; Suwanpakdee, P.; Rujkijyanont, P.; Charoenkwan, P. Clinical Outcomes and Diagnostic Consistency of Serum and CSF Tumor Markers in Pediatric Intracranial Germ Cell Tumors in Thailand: A Multicenter Study. Glob. Pediatr. Health 2022, 9, 2333794X221141765. [Google Scholar] [CrossRef]
- Legault, G.; Allen, J.C. Potential role of ventricular tumor markers in CNS germ cell tumors. Pediatr. Blood Cancer 2013, 60, 1647–1650. [Google Scholar] [CrossRef]
- Takami, H.; Graffeo, C.S.; Perry, A.; Giannini, C.; Nakazato, Y.; Saito, N.; Matsutani, M.; Nishikawa, R.; Ichimura, K.; Daniels, D.J. Roles of Tumor Markers in Central Nervous System Germ Cell Tumors Revisited with Histopathology-Proven Cases in a Large international cohort. Cancers 2022, 14, 979. [Google Scholar] [CrossRef]
- Hong, K.T.; Han, J.W.; Fuji, H.; Byun, H.K.; Koh, K.N.; Wong, R.X.; Lee, H.L.; Yoon, H.I.; Lee, J.H.; Phi, J.H.; et al. Outcomes of intracranial non-germinomatous germ cell tumors: A retrospective Asian multinational study on treatment strategies and prognostic factors. J. Neurooncol. 2022, 160, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Abu-Arja, M.H.; Osorio, D.S.; Lassaletta, A.; Graham, R.T.; Coven, S.L.; Stanek, J.R.; Bouffet, E.; Finlay, J.L.; Abdelbaki, M.S. Prognostic factors for patients with relapsed central nervous system nongerminomatous germ cell tumors. Pediatr. Blood Cancer 2022, 69, e29365. [Google Scholar] [CrossRef]
- Cheng, C.M.; Chiang, Y.H.; Nieh, S. Pineal region teratoma with high serum and CSF alpha-fetoprotein levels. J. Clin. Neurosci. 2006, 13, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Niramis, R.; Anuntkosol, M.; Buranakitjaroen, V.; Tongsin, A.; Mahatharadol, V.; Poocharoen, W.; La-Orwong, S.; Tiansri, K. Long-Term Outcomes of Sacrococcygeal Germ Cell Tumors in Infancy and Childhood. Surg. Res. Pract. 2015, 2015, 398549. [Google Scholar] [CrossRef]
- Chen, S.H.; Du, C.J.; Lai, J.Y.; Chang, T.Y.; Yang, C.P.; Hung, I.J.; Jaing, T.H.; Ming, Y.C.; Hsueh, C. Malignant sacrococcygeal germ cell tumors in children in Taiwan: A retrospective single-center case series. Medicine 2021, 100, e24323. [Google Scholar] [CrossRef]
- Phi, J.H. Sacrococcygeal Teratoma: A Tumor at the Center of Embryogenesis. J. Korean Neurosurg. Soc. 2021, 64, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kirkinen, P.; Heinonen, S.; Vanamo, K.; Ryynänen, M. Maternal serum alpha-fetoprotein and epithelial tumour marker concentrations are not increased by fetal sacrococcygeal teratoma. Prenat. Diagn. 1997, 17, 47–50. [Google Scholar] [CrossRef]
- Nogales, F.F.; Preda, O.; Nicolae, A. Yolk sac tumours revisited. A review of their many faces and names. Histopathology. 2012, 60, 1023–1033. [Google Scholar] [CrossRef]
- Shah, J.P.; Kumar, S.; Bryant, C.S.; Ali-Fehmi, R.; Malone, J.M., Jr.; Deppe, G.; Morris, R.T. A population-based analysis of 788 cases of yolk sac tumors: A comparison of males and females. Int. J. Cancer 2008, 123, 2671–2675. [Google Scholar] [CrossRef]
- Ma, X.; Cao, D.; Peng, P.; Xiao, Y.; Yang, J.; Huang, H.; Zhang, Y.; Yu, M.; Wang, J.; Zhou, H.; et al. Preservation of sexual and reproductive function in the treatment of extragonadal yolk sac tumors in the female genital tract. Front. Pediatr. 2022, 10, 1004501. [Google Scholar] [CrossRef]
- Xiao, G.Q.; Priemer, D.S.; Wei, C.; Aron, M.; Yang, Q.; Idrees, M.T. ZBTB16 is a sensitive and specific marker in detection of metastatic and extragonadal yolk sac tumour. Histopathology 2017, 71, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Littooij, A.S.; McHugh, K.; McCarville, M.B.; Sebire, N.J.; Bahrami, A.; Roebuck, D.J. Yolk sac tumour: A rare cause of raised serum alpha-foetoprotein in a young child with a large liver mass. Pediatr. Radiol. 2014, 44, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Qayoom, S.; Shabbir, N.; Singhai, A.; Verma, N.; Rawat, S. Primary Pure Intrarenal Yolk Sac Tumor in 1.5-Year-Old Boy-A Rare Case Report. Int. J. Surg. Pathol. 2023, 10668969221149131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Xia, C.; Yang, J.; Liu, Z.; Zhao, X.; Li, Y.; Liu, B.; Yang, Y.; She, Y. Renal Yolk Sac Tumor Clinically Misdiagnosed as Nephroblastoma: A Case Report. Fetal Pediatr. Pathol. 2023, 42, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, C.; Orazi, A.; Massimino, M.; Luksch, R. A cystic partially differentiated nephroblastoma producing alpha-fetoprotein. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Crocoli, A.; Madafferi, S.; Jenkner, A.; Zaccara, A.; Inserra, A. Elevated serum alpha-fetoprotein in Wilms tumor may follow the same pattern of other fetal neoplasms after treatment: Evidence from three cases. Pediatr. Surg. Int. 2008, 24, 499–502. [Google Scholar] [CrossRef]
- Yang, A.; Patterson, A.; Pavlock, T.; Chen, K.S.; Gagan, J.; Hatley, M.E.; Frazier, A.L.; Amatruda, J.F.; Laetsch, T.W.; Rakheja, D. Pitfalls in the diagnosis of yolk sac tumor: Lessons from a clinical trial. Pediatr. Blood Cancer 2022, 69, e29451. [Google Scholar] [CrossRef]
- Guo, Y.L.; Zhang, Y.L.; Zhu, J.Q. Prognostic value of serum α-fetoprotein in ovarian yolk sac tumors: A systematic review and meta-analysis. Mol. Clin. Oncol. 2015, 3, 125–132. [Google Scholar] [CrossRef] [PubMed]
- de la Motte Rouge, T.; Pautier, P.; Genestie, C.; Rey, A.; Gouy, S.; Leary, A.; Haie-Meder, C.; Kerbrat, P.; Culine, S.; Fizazi, K.; et al. Prognostic significance of an early decline in serum alpha-fetoprotein during chemotherapy for ovarian yolk sac tumors. Gynecol. Oncol. 2016, 142, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Paramita, P.; Preeti, A.; Mili, J.; Ridhi, J.; Mala, S.; Mm, G. Spectrum of Germ Cell Tumor (GCT): 5 Years’ Experience in a Tertiary Care Center and Utility of OCT4 as a Diagnostic Adjunct. Indian J. Surg. Oncol. 2022, 13, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Raynald; Yang, H.; Wang, J.; Du, J.; Zhang, W.; Shao, Q.; Li, C. Primary intracranial embryonal carcinoma in children: Report of two cases with review of the literature. Int. J. Clin. Exp. Pathol. 2017, 10, 10700–10710. [Google Scholar]
- Shu, H.; Yang, X.H.; Gao, A.F. Ovarian Sertoli-Leydig cell tumor in a 9-month-old infant with special histologic pattern. Fetal Pediatr. Pathol. 2012, 31, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Mooney, E.E.; Nogales, F.F.; Tavassoli, F.A. Hepatocytic differentiation in retiform Sertoli-Leydig cell tumors: Distinguishing a heterologous element from Leydig cells. Hum. Pathol. 1999, 30, 611–617. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; Al-Othman, Y.; Hijazi, E.; McCluggage, W.G. A Report of Ovarian Sertoli-Leydig Cell Tumors With Heterologous Intestinal-type Glands and Alpha Fetoprotein Elevation and Review of the Literature. Int. J. Gynecol. Pathol. 2018, 37, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Strus, M.; Rajtar-Ciosek, A.; Jach, R.; Hankus, J.; Szczepański, W. Ovarian Sertoli-Leydig cell tumour with α-fetoprotein-producing intestinal glandular cells. Clinical case and short review of basic literature. Pol. J. Pathol. 2019, 70, 226–231. [Google Scholar] [CrossRef]
- Serife, K.; Karampelas, S.; Hottat, N.; Devalck, C.; Vanden Houte, K. 15-Year-Old Patient with an Unusual Alpha-Fetoprotein-Producing Sertoli-Leydig Cell Tumor of Ovary. Case Rep. Obstet. Gynecol. 2022, 2022, 4759826. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
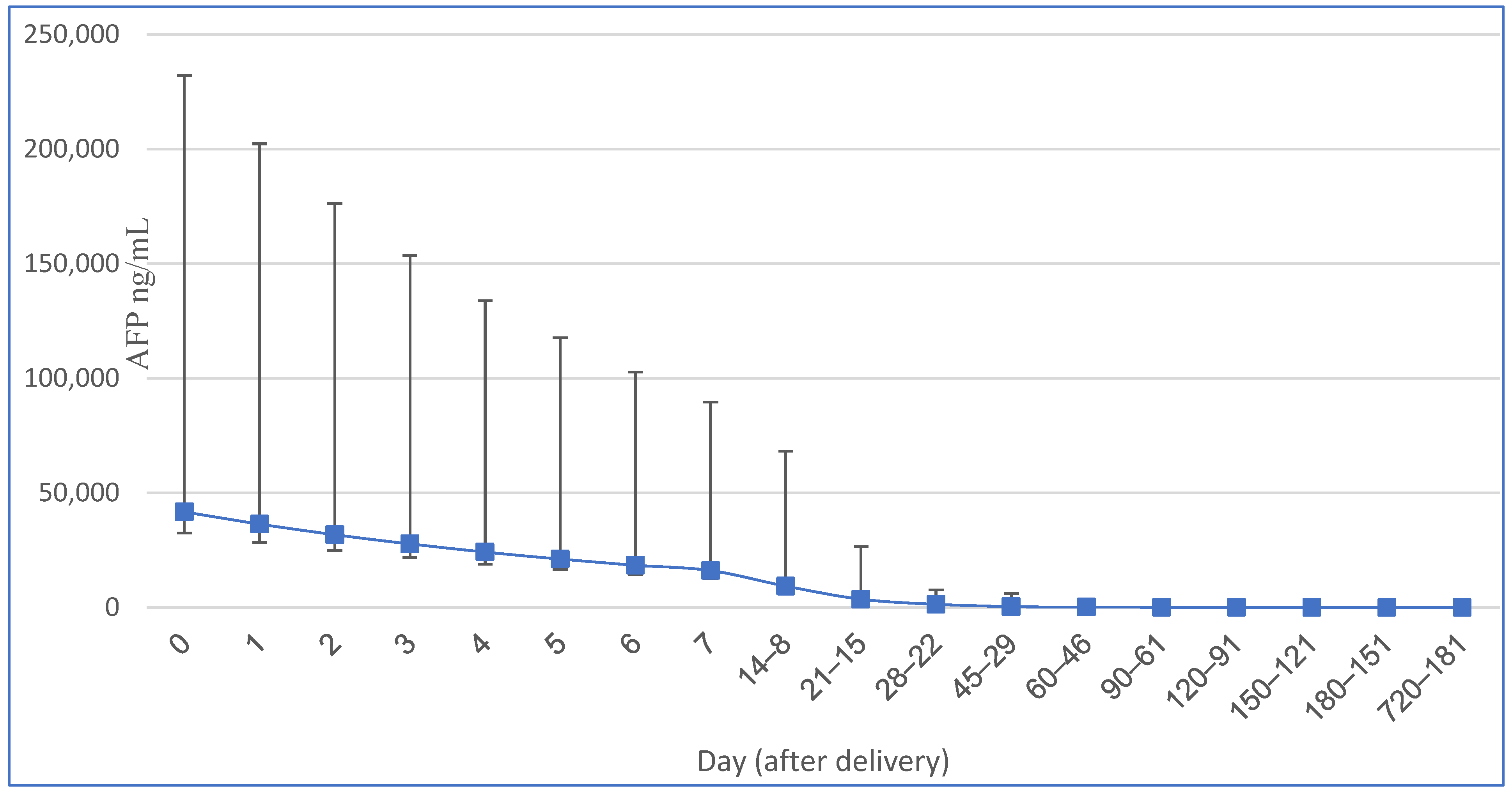
| Neonatal Age (Days) | AFP Mean (ng/mL) | AFP 95.5% Interval (ng/mL) | Half-Life (Days) |
|---|---|---|---|
| 0 | 41,687 | 9120–190,546 | |
| 1 | 36,391 | 7943–165,959 | |
| 2 | 31,769 | 6950–144,544 | |
| 3 | 27,733 | 6026–125,893 | |
| 4 | 24,210 | 5297–109,648 | |
| 5 | 21,135 | 4624–96,605 | 5.1 |
| 6 | 18,450 | 4037–84,334 | 5.1 |
| 7 | 16,107 | 3524–73,621 | 5.1 |
| 8–14 | 9333 | 1480–58,887 | 5.1 |
| 15–21 | 3631 | 575–22,910 | 5.1 |
| 22–28 | 1396 | 316–6310 | 5.1 |
| 29–45 | 417 | 30–5754 | 14 |
| 46–60 | 178 | 16–1995 | 14 |
| 61–90 | 80 | 6–1045 | 28 |
| 91–120 | 36 | 3–417 | 28 |
| 121–150 | 20 | 2–216 | 42 |
| 151–180 | 13 | 1.25–129 | 42 |
| 181–720 | 8 | 0.8–87 | no correlation |
| Risk Factor with Elevated AFP | Risk Factor with Decreased or Normal AFP |
|---|---|
| Hepatitis B | Alpha-1 antitrypsin deficiency |
| Tyrosinemia | Glycogenosis |
| Progressive familiar intrahepatic cholestasis type 2 | Parto-systematic shunts |
| Transaldolase deficiency | Alagille syndrome |
| Ataxia teleangiectasia | Gardner syndrome |
| Fanconi anemia | Familial adenomatous polyposis |
| Biliary artesia | Budd–Chiari syndrome |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Głowska-Ciemny, J.; Szymanski, M.; Kuszerska, A.; Rzepka, R.; von Kaisenberg, C.S.; Kocyłowski, R. Role of Alpha-Fetoprotein (AFP) in Diagnosing Childhood Cancers and Genetic-Related Chronic Diseases. Cancers 2023, 15, 4302. https://doi.org/10.3390/cancers15174302
Głowska-Ciemny J, Szymanski M, Kuszerska A, Rzepka R, von Kaisenberg CS, Kocyłowski R. Role of Alpha-Fetoprotein (AFP) in Diagnosing Childhood Cancers and Genetic-Related Chronic Diseases. Cancers. 2023; 15(17):4302. https://doi.org/10.3390/cancers15174302
Chicago/Turabian StyleGłowska-Ciemny, Joanna, Marcin Szymanski, Agata Kuszerska, Rafał Rzepka, Constantin S. von Kaisenberg, and Rafał Kocyłowski. 2023. "Role of Alpha-Fetoprotein (AFP) in Diagnosing Childhood Cancers and Genetic-Related Chronic Diseases" Cancers 15, no. 17: 4302. https://doi.org/10.3390/cancers15174302
APA StyleGłowska-Ciemny, J., Szymanski, M., Kuszerska, A., Rzepka, R., von Kaisenberg, C. S., & Kocyłowski, R. (2023). Role of Alpha-Fetoprotein (AFP) in Diagnosing Childhood Cancers and Genetic-Related Chronic Diseases. Cancers, 15(17), 4302. https://doi.org/10.3390/cancers15174302