

Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients
 , , ,
, , ,  ,
,  , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
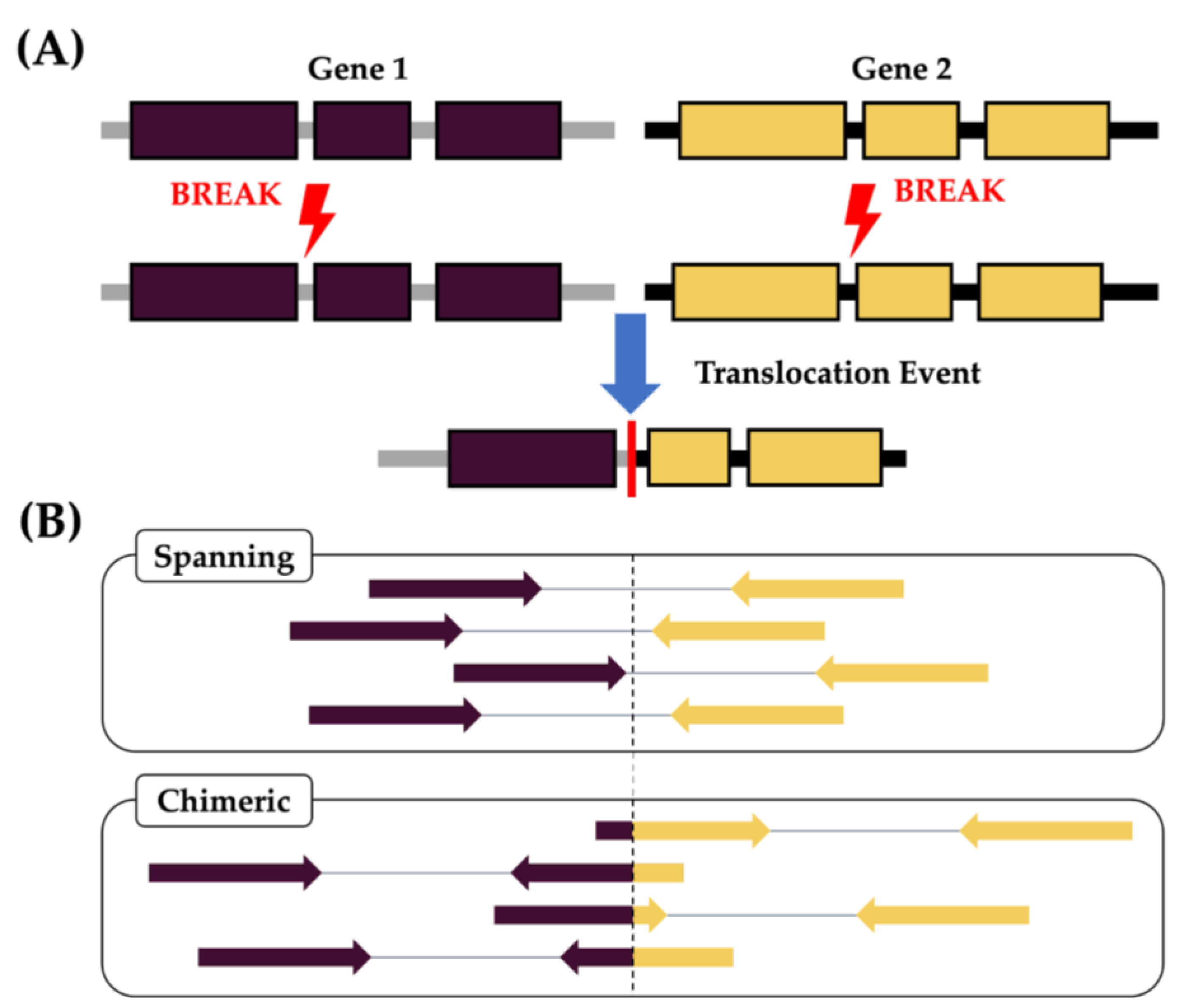
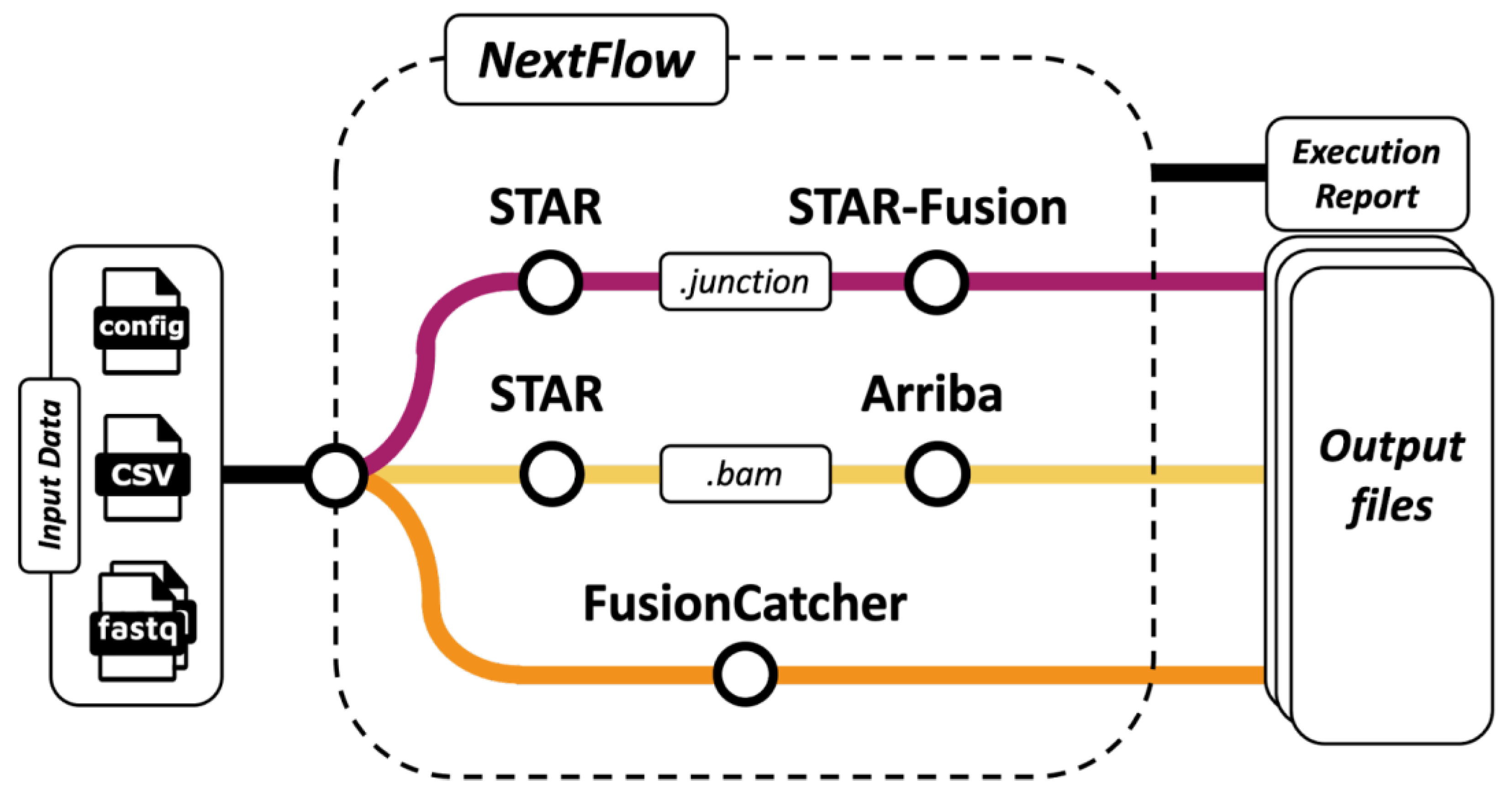
2. Materials and Methods
3. Results
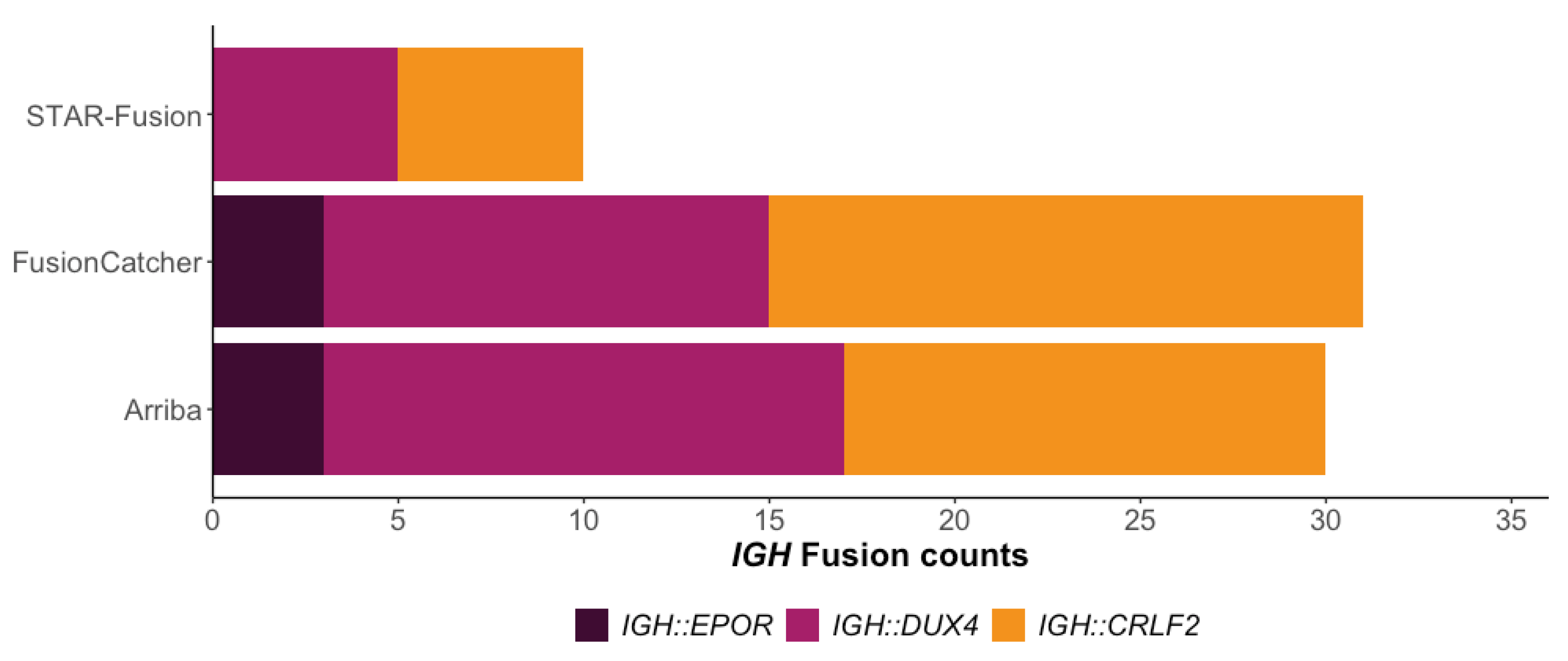
3.1. Using RIGHT to Detect IGH Gene Fusion Events
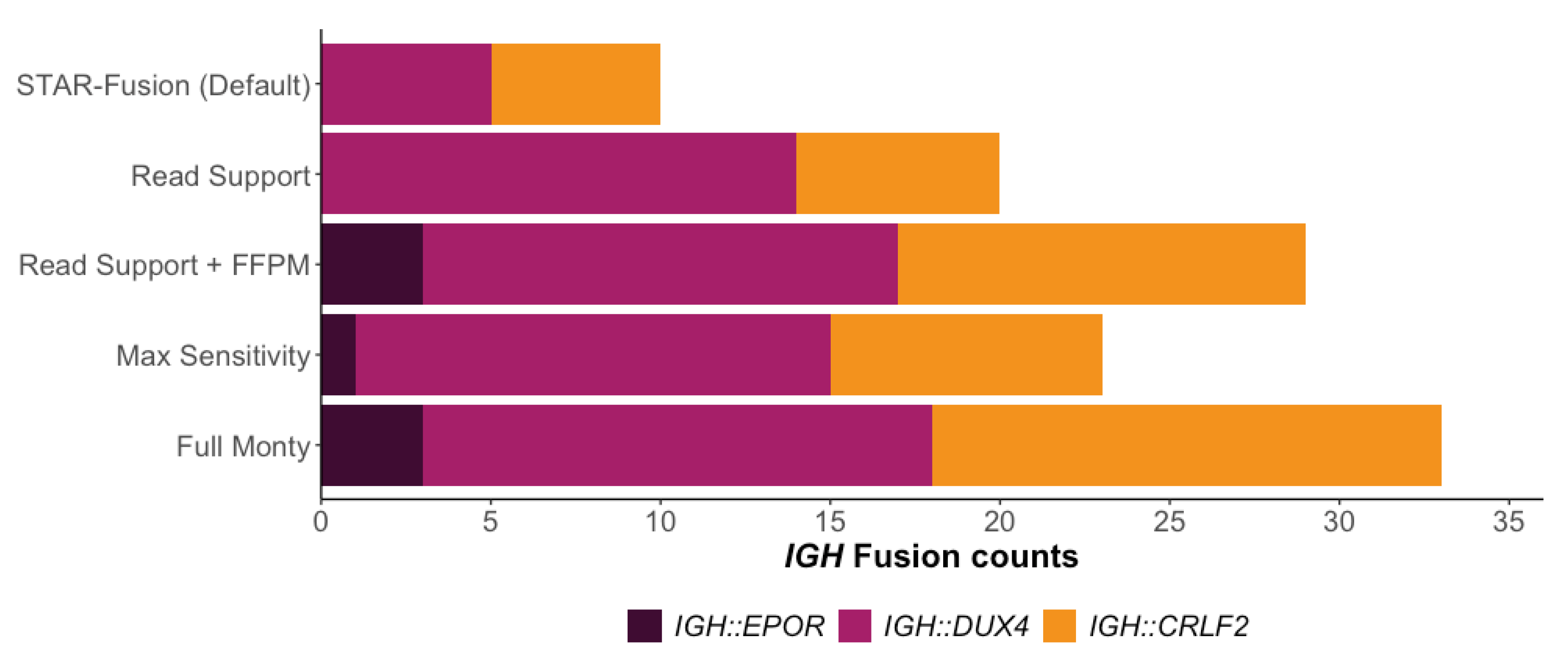
3.2. Improving STAR-Fusions Performance when Detecting IGH Gene Fusions
3.3. Runtime and Resources
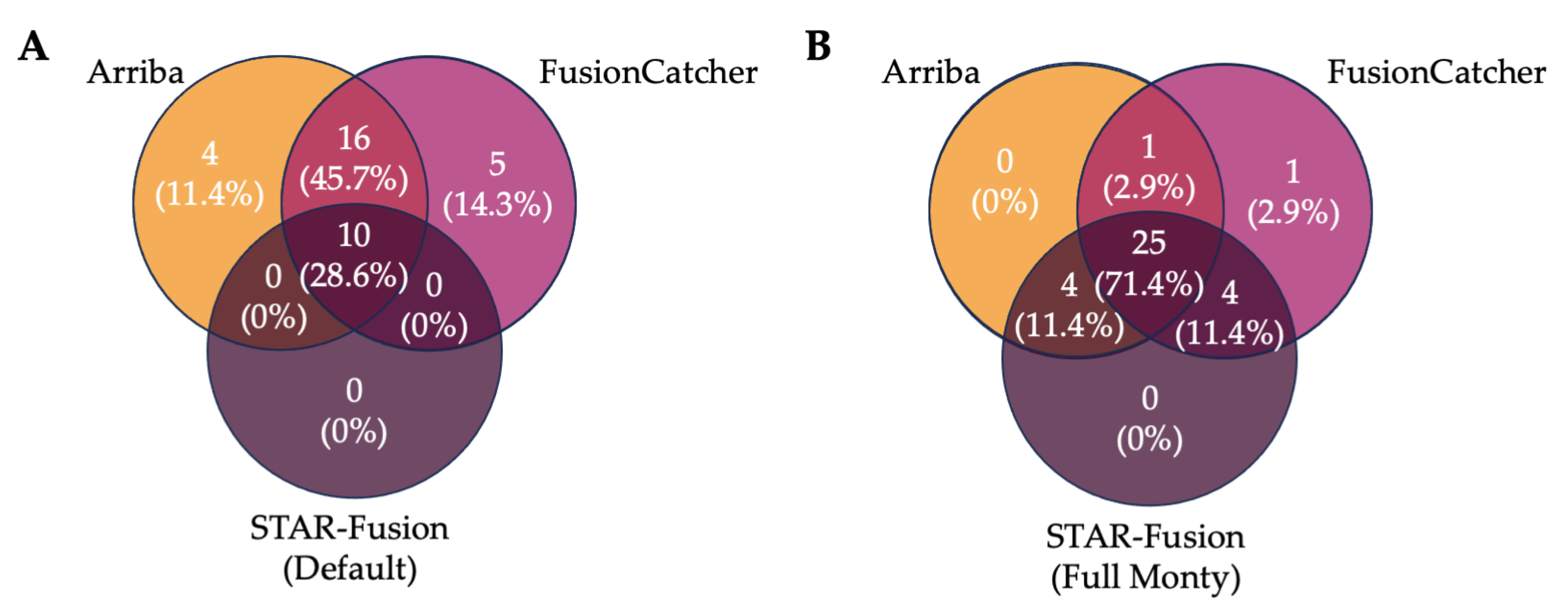
3.4. Ensemble Filtering Approaches to Identify IGH Fusions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G. Genetics and prognosis of ALL in children vs. adults. Hematology 2018, 2018, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Australian Institute of Health and Welfare (AIHW). Cancer Data in Australia. 2022. Available online: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/data?page=3 (accessed on 25 August 2022).
- Russell, L.J.; Enshaei, A.; Jones, L.; Erhorn, A.; Masic, D.; Bentley, H.; Laczko, K.S.; Fielding, A.K.; Goldstone, A.H.; Goulden, N.; et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J. Clin. Oncol. 2014, 32, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Harrison, C.J. Advances in B-cell precursor acute lymphoblastic leukemia genomics. HemaSphere 2018, 2, e53. [Google Scholar] [CrossRef]
- Schröder, J.; Kumar, A.; Wong, S.Q. Overview of Fusion Detection Strategies Using Next-Generation Sequencing. Methods Mol. Biol. 2019, 1908, 125–138. [Google Scholar]
- Ryan, S.L.; Peden, J.F.; Kingsbury, Z.; Schwab, C.J.; James, T.; Polonen, P.; Mijuskovic, M.; Becq, J.; Yim, R.; Cranston, R.E.; et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia 2023, 37, 518–528. [Google Scholar] [CrossRef]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; Saint-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022, 6, 1329–1341. [Google Scholar] [CrossRef]
- Kim, J.C.; Zuzarte, P.C.; Murphy, T.; Chan-Seng-Yue, M.; Brown, A.M.K.; Krzyzanowski, P.M.; Smith, A.C.; Notta, F.; Minden, M.D.; McPherson, J.D. Cryptic genomic lesions in adverse-risk acute myeloid leukemia identified by integrated whole genome and transcriptome sequencing. Leukemia 2020, 34, 306–311. [Google Scholar] [CrossRef]
- Coudray, A.; Battenhouse, A.M.; Bucher, P.; Iyer, V.R. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. PeerJ 2018, 6, e5362. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Breden, F. The immunoglobulin heavy chain locus: Genetic variation, missing data, and implications for human disease. Genes Immun. 2012, 13, 363–373. [Google Scholar] [CrossRef]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 1, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Giallourakis, C.; Mostoslavsky, R.; Alt, F.W. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu. Rev. Immunol. 2006, 24, 541–570. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Gadala-Maria, D.; Gidoni, M.; Marquez, S.; Vander Heiden, J.A.; Kos, J.T.; Watson, C.T.; O’Connor, K.C.; Yaari, G.; Kleinstein, S.H. Identification of Subject-Specific Immunoglobulin Alleles From Expressed Repertoire Sequencing Data. Front. Immunol. 2019, 10, 129. [Google Scholar] [CrossRef]
- Jackson, M.; Kavoussanakis, K.; Wallace, E.W.J. Using prototyping to choose a bioinformatics workflow management system. PLoS Comput. Biol. 2021, 17, e1008622. [Google Scholar] [CrossRef]
- Chapiro, E.; Radford-Weiss, I.; Cung, H.-A.; Dastugue, N.; Nadal, N.; Taviaux, S.; Barin, C.; Struski, S.; Talmant, P.; Vandenberghe, P.; et al. Chromosomal translocations involving the IGH@ locus in B-cell precursor acute lymphoblastic leukemia: 29 new cases and a review of the literature. Cancer Genet. 2013, 206, 162–173. [Google Scholar] [CrossRef]
- Russell, L.J.; Jones, L.; Erhorn, A.; Ensahei, A.; Masic, D.; Bentley, H.; Laczko, K.S.; Harrison, C.J. Immunoglobulin Heavy Chain Locus (IGH@) Translocations in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL): Incidence and Risk Stratification. Blood 2012, 120, 1274. [Google Scholar] [CrossRef]
- Dyer, M.J.S.; Akasaka, T.; Capasso, M.; Dusanjh, P.; Lee, Y.F.; Karran, E.L.; Nagel, I.; Vater, I.; Cario, G.; Siebert, R. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: Rare clinical curios or potent genetic drivers? Blood 2010, 115, 1490–1499. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Wratten, L.; Wilm, A.; Göke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat. Methods 2021, 18, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Spišaková, V.; Hejtmánek, L.; Hynšt, J. Nextflow in bioinformatics: Executors performance comparison using genomics data. Future Gener. Comput. Syst. 2023, 142, 328–339. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014, 011650. [Google Scholar] [CrossRef]
- Genome Reference Consortium. Available online: https://www.ncbi.nlm.nih.gov/grc (accessed on 15 July 2022).
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.-C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.; et al. Modernizing reference genome assemblies. PLoS Biol. 2011, 9, e1001091. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Rehn, J.; Mayoh, C.; Heatley, S.L.; McClure, B.J.; Eadie, L.N.; Schutz, C.; Yeung, D.T.; Cowley, M.J.; Breen, J.; White, D.L. RaScALL: Rapid (Ra) screening (Sc) of RNA-seq data for prognostically significant genomic alterations in acute lymphoblastic leukaemia (ALL). PLoS Genet. 2022, 18, e1010300. [Google Scholar] [CrossRef] [PubMed]
- RaScALL: Rapid Screening of RNA-Seq in Acute Lymphoblastic Leukaemia—EGA European Genome-Phenome Archive. Available online: https://ega-archive.org/studies/EGAS00001006460 (accessed on 27 February 2023).
- Selvam, P.; Kelly, K.; Hesse, A.N.; Spitzer, D.; Reddi, H.V. Evaluating gene fusions in solid tumors—Clinical experience using an RNA based 53 gene next-generation sequencing panel. Cancer Genet. 2019, 233–234, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Painuli, D.; Bhardwaj, S.; Köse, U. Recent advancement in cancer diagnosis using machine learning and deep learning techniques: A comprehensive review. Comput. Biol. Med. 2022, 146, 105580. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.; Singh, A.; Bhalla, V.K. A Systematic Review on Biomarker Identification for Cancer Diagnosis and Prognosis in Multi-omics: From Computational Needs to Machine Learning and Deep Learning. Arch. Comput. Methods Eng. 2023, 30, 917–949. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Naquin, D.; Gorrichon, K.; Jaszczyszyn, Y.; Ouazahrou, R.; Thermes, C.; Hernandez, C. Genomics in the long-read sequencing era. Trends Genet. 2023. [Google Scholar] [CrossRef]
- Tian, L.; Shao, Y.; Nance, S.; Dang, J.; Xu, B.; Ma, X.; Li, Y.; Ju, B.; Dong, L.; Newman, S.; et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Nat. Commun. 2019, 10, 2789. [Google Scholar] [CrossRef]
- Tian, L.; Li, Y.; Edmonson, M.N.; Zhou, X.; Newman, S.; McLeod, C.; Thrasher, A.; Liu, Y.; Tang, B.; Rusch, M.C.; et al. CICERO: A versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol. 2020, 21, 126. [Google Scholar] [CrossRef]
- Davidson, N.M.; Chen, Y.; Sadras, T.; Ryland, G.L.; Blombery, P.; Ekert, P.G.; Göke, J.; Oshlack, A. JAFFAL: Detecting fusion genes with long-read transcriptome sequencing. Genome Biol. 2022, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Malouf, G. P482: Accufusion: A Highly Scalable Software Tool for Detecting Gene Fusions By Rna-Seq in Leukemia. HemaSphere 2023, 7. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | RSS (GB) | |||||
|---|---|---|---|---|---|---|
| Max | Mean | Min | Max | Mean | Min | |
| Arriba | 27 m 44 s | 15 m | 6 m 43 s | 38.2 | 35.8 | 34.5 |
| FusionCatcher | 4 h 25 m 21 s | 2 h 39 m | 1 h 23 m 50 s | 27.5 | 24.9 | 22.5 |
| STAR-Fusion (Default) | 1 h 38 m 6 s | 55 m | 27 m 37 s | 39.2 | 38.3 | 37.5 |
| Read Support | 1 h 33 m 6 s | 57 m | 26 m 35 s | 39.2 | 38.3 | 37.4 |
| Read Support + FFPM | 1 h 29 m 18 s | 56 m | 27 m 4 s | 39.2 | 38.3 | 37.4 |
| Max Sensitivity | 2 h 11 m 36 s | 1 h 3 m | 26 m 21 s | 39.2 | 38.3 | 37.5 |
| Full Monty | 2 h 11 s | 1 h 3 m | 26 m 52 s | 39.2 | 38.5 | 37.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomson, A.J.; Rehn, J.A.; Heatley, S.L.; Eadie, L.N.; Page, E.C.; Schutz, C.; McClure, B.J.; Sutton, R.; Dalla-Pozza, L.; Moore, A.S.; et al. Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients. Cancers 2023, 15, 4731. https://doi.org/10.3390/cancers15194731
Thomson AJ, Rehn JA, Heatley SL, Eadie LN, Page EC, Schutz C, McClure BJ, Sutton R, Dalla-Pozza L, Moore AS, et al. Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients. Cancers. 2023; 15(19):4731. https://doi.org/10.3390/cancers15194731
Chicago/Turabian StyleThomson, Ashlee J., Jacqueline A. Rehn, Susan L. Heatley, Laura N. Eadie, Elyse C. Page, Caitlin Schutz, Barbara J. McClure, Rosemary Sutton, Luciano Dalla-Pozza, Andrew S. Moore, and et al. 2023. "Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients" Cancers 15, no. 19: 4731. https://doi.org/10.3390/cancers15194731
APA StyleThomson, A. J., Rehn, J. A., Heatley, S. L., Eadie, L. N., Page, E. C., Schutz, C., McClure, B. J., Sutton, R., Dalla-Pozza, L., Moore, A. S., Greenwood, M., Kotecha, R. S., Fong, C. Y., Yong, A. S. M., Yeung, D. T., Breen, J., & White, D. L. (2023). Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients. Cancers, 15(19), 4731. https://doi.org/10.3390/cancers15194731