

Selective Activation of M1 Muscarinic Receptors Attenuates Human Colon Cancer Cell Proliferation
 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Lines and Cell Culture
2.3. Human Tissues
2.4. Immunoblot Analysis
2.5. Quantitative Real-Time PCR (qPCR)
2.6. Cell Proliferation Assay
2.7. Lactate Dehydrogenase (LDH) Release Assay
2.8. Senescence-Associated β-Galactosidase
2.9. Statistical Analysis
3. Results
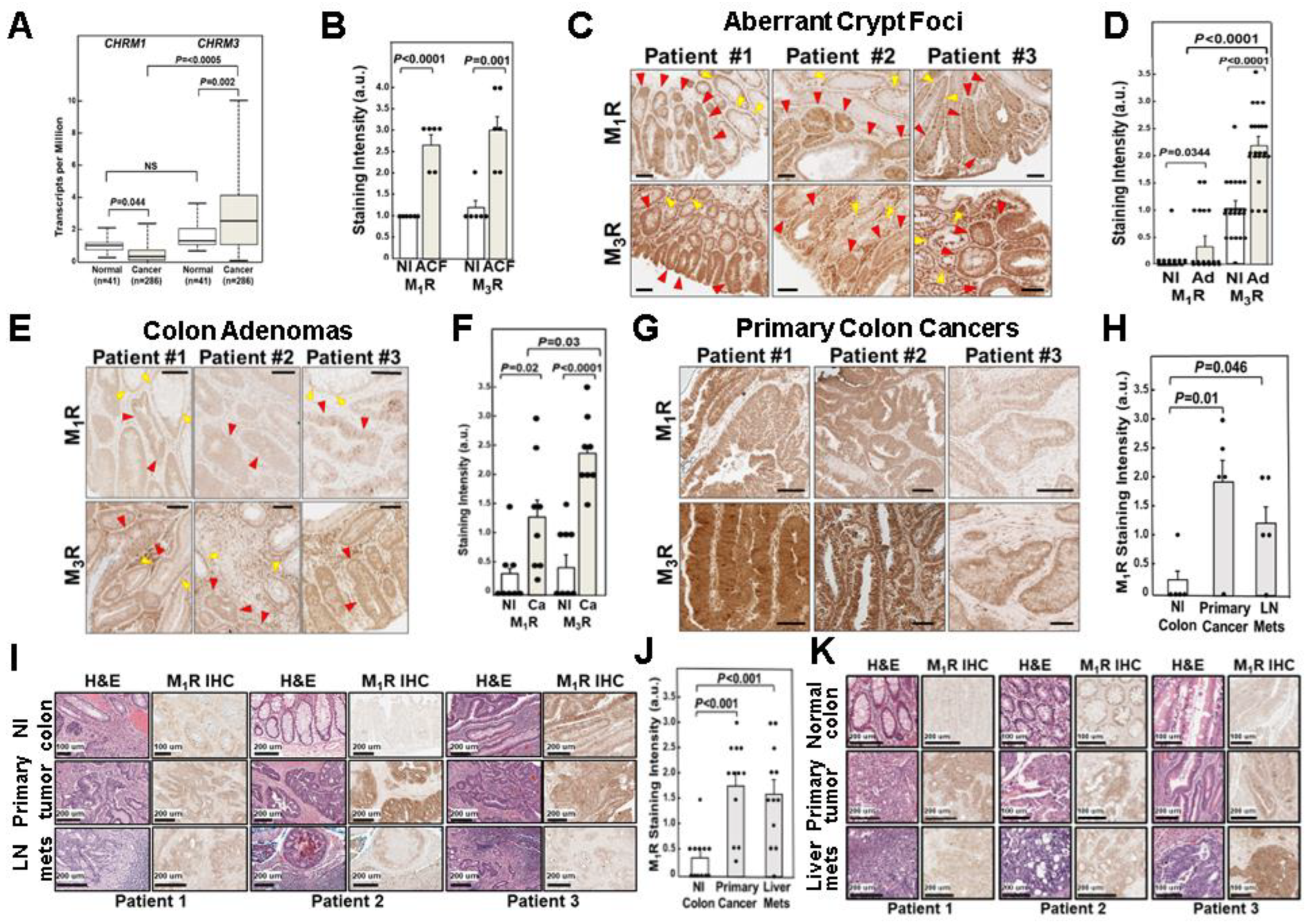
3.1. Relative CHRM1 and CHRM3 mRNA Expression in Colorectal Adenocarcinomas
3.2. Relative M1R and M3R Protein Expression in Progressive Colon Neoplasia
3.2.1. Relative M1R and M3R Protein Expression in Aberrant Crypt Foci
3.2.2. Relative M1R and M3R Protein Expression in Colon Adenomas
3.2.3. Relative M1R and M3R Protein Expression in Primary Colon Adenocarcinomas
3.2.4. M1R Protein Expression in Colon Cancer Lymph Node and Liver Metastases
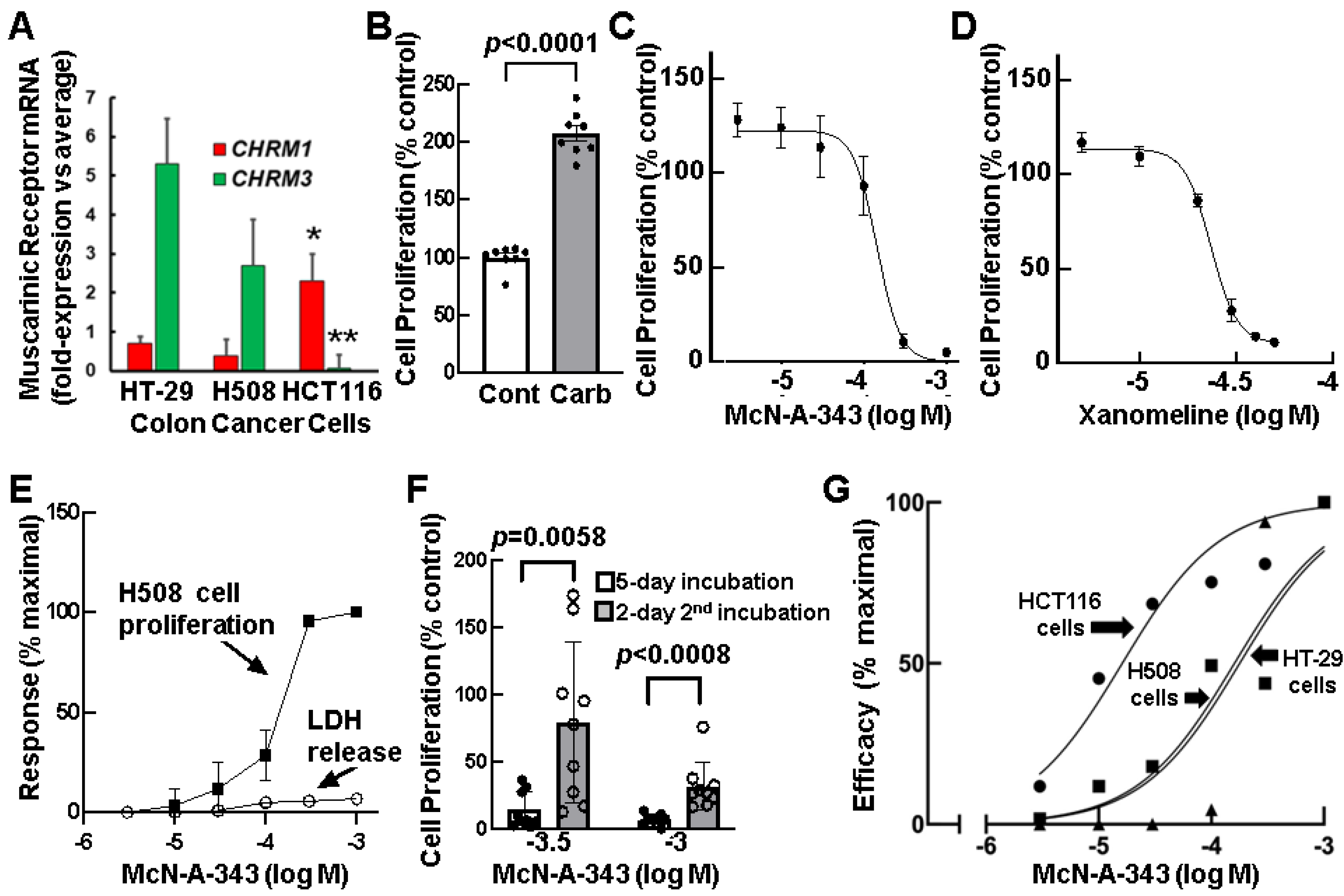
3.3. Relative Expression of CHRM1 and CHRM3 in Human Colon Cancer Cells
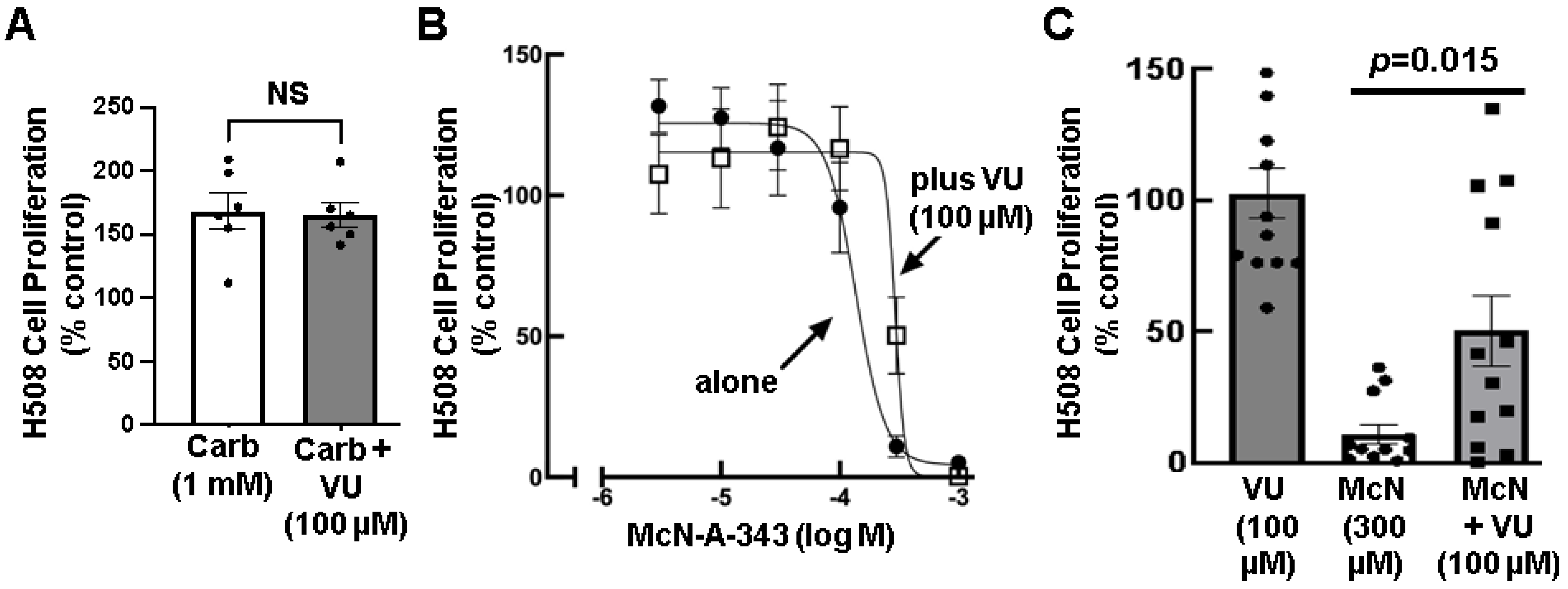
3.4. Effects of Selective M1R Activation on Human Colon Cancer Cell Proliferation
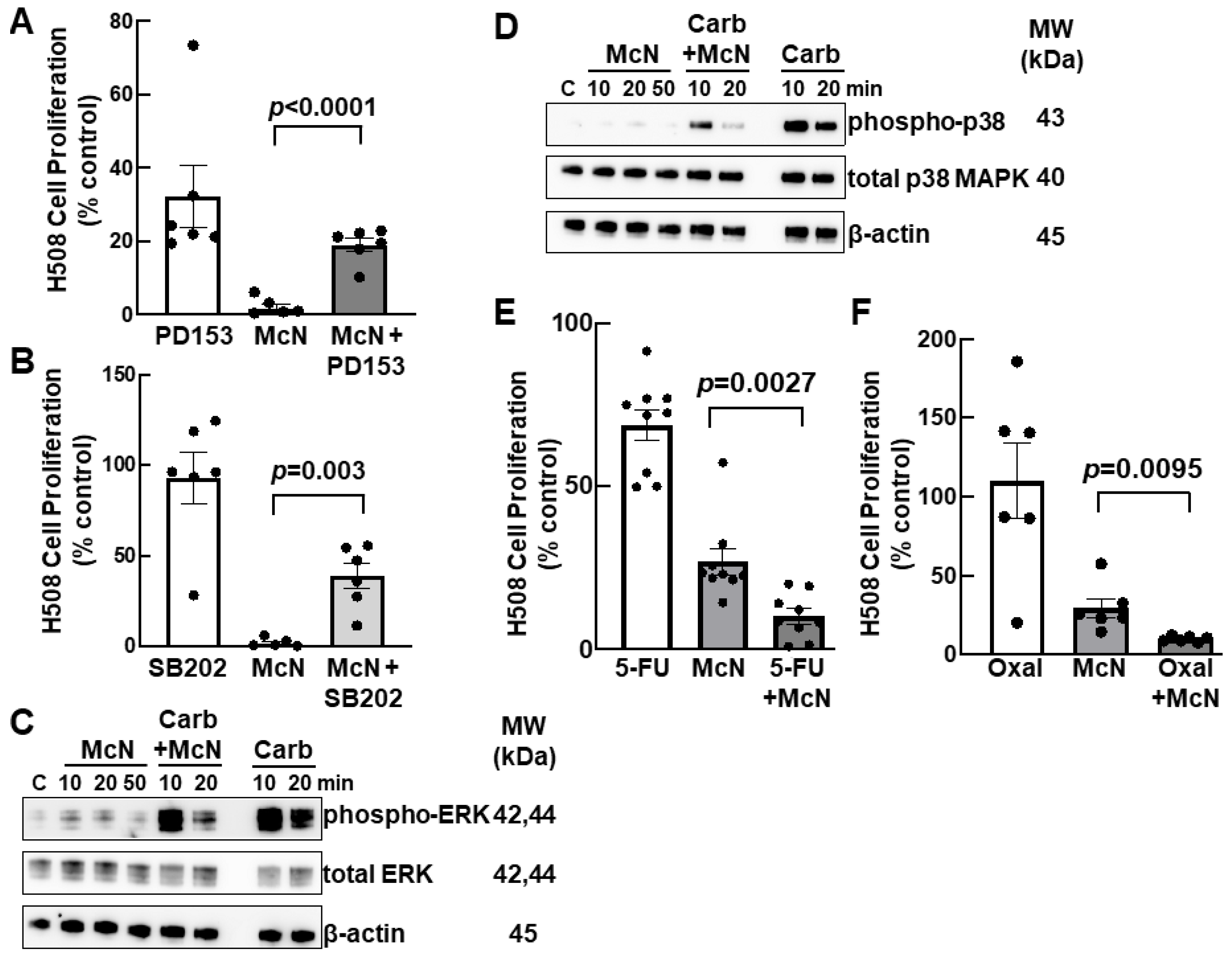
3.5. Inhibiting EGFR and p38 MAPK Activation Attenuates the Inhibitory Actions of an M1R Agonist on Colon Cancer Cell Proliferation
3.6. Combining an M1R Agonist with Conventional Colon Cancer Chemotherapy Enhances Inhibition of Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Middelhoff, M.; Nienhuser, H.; Valenti, G.; Maurer, H.C.; Hayakawa, Y.; Takahashi, R.; Kim, W.; Jiang, Z.; Malagola, E.; Cuti, K.; et al. Prox1-positive cells monitor and sustain the murine intestinal epithelial cholinergic niche. Nat. Commun. 2020, 11, 111. [Google Scholar] [CrossRef]
- Cheng, K.; Shang, A.C.; Drachenberg, C.B.; Zhan, M.; Raufman, J.P. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 2017, 8, 21106–21114. [Google Scholar] [CrossRef]
- Alizadeh, M.; Schledwitz, A.; Cheng, K.; Raufman, J.P. Mechanistic Clues Provided by Concurrent Changes in the Expression of Genes Encoding the M(1) Muscarinic Receptor, beta-Catenin Signaling Proteins, and Downstream Targets in Adenocarcinomas of the Colon. Front. Physiol. 2022, 13, 857563. [Google Scholar] [CrossRef]
- Cheng, K.; Xie, G.; Khurana, S.; Heath, J.; Drachenberg, C.B.; Timmons, J.; Shah, N.; Raufman, J.P. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol. Cancer 2014, 13, 77. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res. 2007, 67, 3936–3944. [Google Scholar] [CrossRef]
- Frucht, H.; Jensen, R.T.; Dexter, D.; Yang, W.-L.; Xiao, Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin. Cancer Res. 1999, 5, 2532–2539. [Google Scholar]
- Yang, W.L.; Frucht, H. Cholinergic receptor up-regulates COX-2 expression and prostaglandin E(2) production in colon cancer cells. Carcinogenesis 2000, 21, 1789–1793. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Abreu Rde, S.; Ko, D.; Le, S.Y.; Shapiro, B.A.; Burns, S.C.; Sandhu, D.; Boutz, D.R.; Marcotte, E.M.; Penalva, L.O. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Michel, M.C.; Wieland, T.; Tsujimoto, G. How reliable are G-protein-coupled receptor antibodies? Naunyn. Schmiedebergs. Arch. Pharmacol. 2009, 379, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Jositsch, G.; Papadakis, T.; Haberberger, R.V.; Wolff, M.; Wess, J.; Kummer, W. Suitability of muscarinic acetylcholine receptor antibodies for immunohistochemistry evaluated on tissue sections of receptor gene-deficient mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2009, 379, 389–395. [Google Scholar] [CrossRef]
- Olianas, M.C.; Dedoni, S.; Onali, P. Involvement of store-operated Ca(2+) entry in activation of AMP-activated protein kinase and stimulation of glucose uptake by M3 muscarinic acetylcholine receptors in human neuroblastoma cells. Biochim. Biophys. Acta. 2014, 1843, 3004–3017. [Google Scholar] [CrossRef]
- Bird, R.P. Observation and quantification of aberrant cruypts in the murine colon treated with a colon carcinogen: Preliminary findings. Cancer Lett. 1987, 37, 147–151. [Google Scholar] [CrossRef]
- Bird, R.P. Role of aberrant crypt foci in understanding the pathogenesis of colon cancer. Cancer Lett. 1995, 93, 55–71. [Google Scholar] [CrossRef]
- Takayama, T.; Katsuki, S.; Takahashi, Y.; Ohi, M.; Nojiri, S.; Sakamaki, S.; Kato, J.; Kogawa, K.; Miyake, H.; Niitsu, Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 1998, 339, 1277–1284. [Google Scholar] [CrossRef]
- Alrawi, S.J.; Schiff, M.; Carroll, R.E.; Dayton, M.; Gibbs, J.F.; Kulavlat, M.; Tan, D.; Berman, K.; Stoler, D.L.; Anderson, G.R. Aberrant crypt foci. Anticancer. Res. 2006, 26, 107–119. [Google Scholar] [PubMed]
- Hawthorn, L.; Lan, L.; Mojica, W. Evidence for field effect cancerization in colorectal cancer. Genomics 2014, 103, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Said, A.H.; Hu, S.; Abutaleb, A.; Watkins, T.; Cheng, K.; Chahdi, A.; Kuppusamy, P.; Saxena, N.; Xie, G.; Raufman, J.P. Interacting post-muscarinic receptor signaling pathways potentiate matrix metalloproteinase-1 expression and invasion of human colon cancer cells. Biochem. J. 2017, 474, 647–665. [Google Scholar] [CrossRef]
- Cheng, K.; Zimniak, P.; Raufman, J.P. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res. 2003, 63, 6744–6750. [Google Scholar] [CrossRef]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef]
- Roszkowski, A.P. An unusual type of sympathetic ganglionic stimulant. J. Pharmacol. Exp. Ther. 1961, 132, 156–170. [Google Scholar]
- Mitchelson, F.J. The pharmacology of McN-A-343. Pharmacol. Ther. 2012, 135, 216–245. [Google Scholar] [CrossRef]
- Eglen, R.M.; Kenny, B.A.; Michel, A.D.; Whiting, R.L. Muscarinic activity of McN-A-343 and its value in muscarinic receptor classification. Br. J. Pharmacol. 1987, 90, 693–700. [Google Scholar] [CrossRef]
- Wakade, A.R.; Kahn, R.; Malhotra, R.K.; Wakade, C.G.; Wakade, T.D. McN-A-343, a specific agonist of M1-muscarinic receptors, exerts antinicotinic and antimuscarinic effects in the rat adrenal medulla. Life Sci. 1986, 39, 2073–2080. [Google Scholar] [CrossRef]
- Shannon, H.E.; Bymaster, F.P.; Calligaro, D.O.; Greenwood, B.; Mitch, C.H.; Sawyer, B.D.; Ward, J.S.; Wong, D.T.; Olesen, P.H.; Sheardown, M.J.; et al. Xanomeline: A novel muscarinic receptor agonist with functional selectivity for M1 receptors. J. Pharmacol. Exp. Ther. 1994, 269, 271–281. [Google Scholar]
- Bender, A.M.; Jones, C.K.; Lindsley, C.W. Classics in Chemical Neuroscience: Xanomeline. ACS Chem. Neurosci. 2017, 8, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.K.; Noetzel, M.J.; De Lorme, K.C.; Jakubik, J.; Dolezal, V.; El-Fakahany, E.E. Pharmacological evaluation of the long-term effects of xanomeline on the M(1) muscarinic acetylcholine receptor. PLoS ONE 2010, 5, e15722. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.; Hissin, P.J.; Propper, W.; Mayer, L.; Sarkozi, L. Stability of lactate dehydrogenase at different storage temperatures. Clin. Biochem. 1986, 19, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Makrodouli, E.; Evagelidou, M.; Joyce, T.; Probert, L.; Pintzas, A. BRAF(V600E) efficient transformation and induction of microsatellite instability versus KRAS(G12V) induction of senescence markers in human colon cancer cells. Neoplasia 2009, 11, 1116–1131. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- O’Donovan, N.; Crown, J.; Stunell, H.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Caspase 3 in breast cancer. Clin. Cancer Res. 2003, 9, 738–742. [Google Scholar]
- Gazi, L.; Bobirnac, I.; Danzeisen, M.; Schupbach, E.; Langenegger, D.; Sommer, B.; Hoyer, D.; Tricklebank, M.; Schoeffter, P. Receptor density as a factor governing the efficacy of the dopamine D4 receptor ligands, L-745,870 and U-101958 at human recombinant D4.4 receptors expressed in CHO cells. Br. J. Pharmacol. 1999, 128, 613–620. [Google Scholar] [CrossRef]
- Robertson, S.; Rohwer, J.M.; Hapgood, J.P.; Louw, A. Impact of glucocorticoid receptor density on ligand-independent dimerization, cooperative ligand-binding and basal priming of transactivation: A cell culture model. PLoS ONE 2013, 8, e64831. [Google Scholar] [CrossRef]
- Koener, B.; Focant, M.C.; Bosier, B.; Maloteaux, J.M.; Hermans, E. Increasing the density of the D2L receptor and manipulating the receptor environment are required to evidence the partial agonist properties of aripiprazole. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 36, 60–70. [Google Scholar] [CrossRef]
- Sheffler, D.J.; Williams, R.; Bridges, T.M.; Xiang, Z.; Kane, A.S.; Byun, N.E.; Jadhav, S.; Mock, M.M.; Zheng, F.; Lewis, L.M.; et al. A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol. Pharmacol. 2009, 76, 356–368. [Google Scholar] [CrossRef]
- Fu, N.; Drinnenberg, I.; Kelso, J.; Wu, J.R.; Paabo, S.; Zeng, R.; Khaitovich, P. Comparison of protein and mRNA expression evolution in humans and chimpanzees. PLoS ONE 2007, 2, e216. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wu, G.; Zhang, W. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: A quantitative analysis. Genetics 2006, 174, 2229–2243. [Google Scholar] [CrossRef] [PubMed]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.S.; Woo, K.K.; Aalderink, M.; Fry, S.; Greenwood, J.M.; Glass, M.; Dragunow, M. M1 muscarinic receptor activation mediates cell death in M1-HEK293 cells. PLoS ONE 2013, 8, e72011. [Google Scholar] [CrossRef]
- Zaccor, N.W.; Sumner, C.J.; Snyder, S.H. The nonselective cation channel TRPV4 inhibits angiotensin II receptors. J. Biol. Chem. 2020, 295, 9986–9997. [Google Scholar] [CrossRef]
- Magalhaes, D.A.; Batista, J.A.; Sousa, S.G.; Ferreira, J.D.S.; da Rocha Rodrigues, L.; Pereira, C.M.C.; do Nascimento Lima, J.V.; de Albuquerque, I.F.; Bezerra, N.; Monteiro, C.; et al. McN-A-343, a muscarinic agonist, reduces inflammation and oxidative stress in an experimental model of ulcerative colitis. Life Sci. 2021, 272, 119194. [Google Scholar] [CrossRef]
- Caccamo, A.; Fisher, A.; LaFerla, F.M. M1 agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr. Alzheimer. Res. 2009, 6, 112–117. [Google Scholar] [CrossRef]
- Brown, A.J.H.; Bradley, S.J.; Marshall, F.H.; Brown, G.A.; Bennett, K.A.; Brown, J.; Cansfield, J.E.; Cross, D.M.; de Graaf, C.; Hudson, B.D.; et al. From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer’s disease. Cell 2021, 184, 5886–5901.e5822. [Google Scholar] [CrossRef]
- Brannan, S.K.; Sawchak, S.; Miller, A.C.; Lieberman, J.A.; Paul, S.M.; Breier, A. Muscarinic Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N. Engl. J. Med. 2021, 384, 717–726. [Google Scholar] [CrossRef]
- Singh, A. Xanomeline and Trospium: A Potential Fixed Drug Combination (FDC) for Schizophrenia-A Brief Review of Current Data. Innov. Clin. Neurosci. 2022, 19, 43–47. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundel, M.H.; Sampaio Moura, N.; Cheng, K.; Chatain, O.; Hu, S.; Drachenberg, C.B.; Xie, G.; Raufman, J.-P. Selective Activation of M1 Muscarinic Receptors Attenuates Human Colon Cancer Cell Proliferation. Cancers 2023, 15, 4766. https://doi.org/10.3390/cancers15194766
Sundel MH, Sampaio Moura N, Cheng K, Chatain O, Hu S, Drachenberg CB, Xie G, Raufman J-P. Selective Activation of M1 Muscarinic Receptors Attenuates Human Colon Cancer Cell Proliferation. Cancers. 2023; 15(19):4766. https://doi.org/10.3390/cancers15194766
Chicago/Turabian StyleSundel, Margaret H., Natalia Sampaio Moura, Kunrong Cheng, Oscar Chatain, Shien Hu, Cinthia B. Drachenberg, Guofeng Xie, and Jean-Pierre Raufman. 2023. "Selective Activation of M1 Muscarinic Receptors Attenuates Human Colon Cancer Cell Proliferation" Cancers 15, no. 19: 4766. https://doi.org/10.3390/cancers15194766
APA StyleSundel, M. H., Sampaio Moura, N., Cheng, K., Chatain, O., Hu, S., Drachenberg, C. B., Xie, G., & Raufman, J. -P. (2023). Selective Activation of M1 Muscarinic Receptors Attenuates Human Colon Cancer Cell Proliferation. Cancers, 15(19), 4766. https://doi.org/10.3390/cancers15194766