
Gene Screening for Prognosis of Non-Muscle-Invasive Bladder Carcinoma under Competing Risks Endpoints


Abstract
:Simple Summary
Abstract
1. Introduction
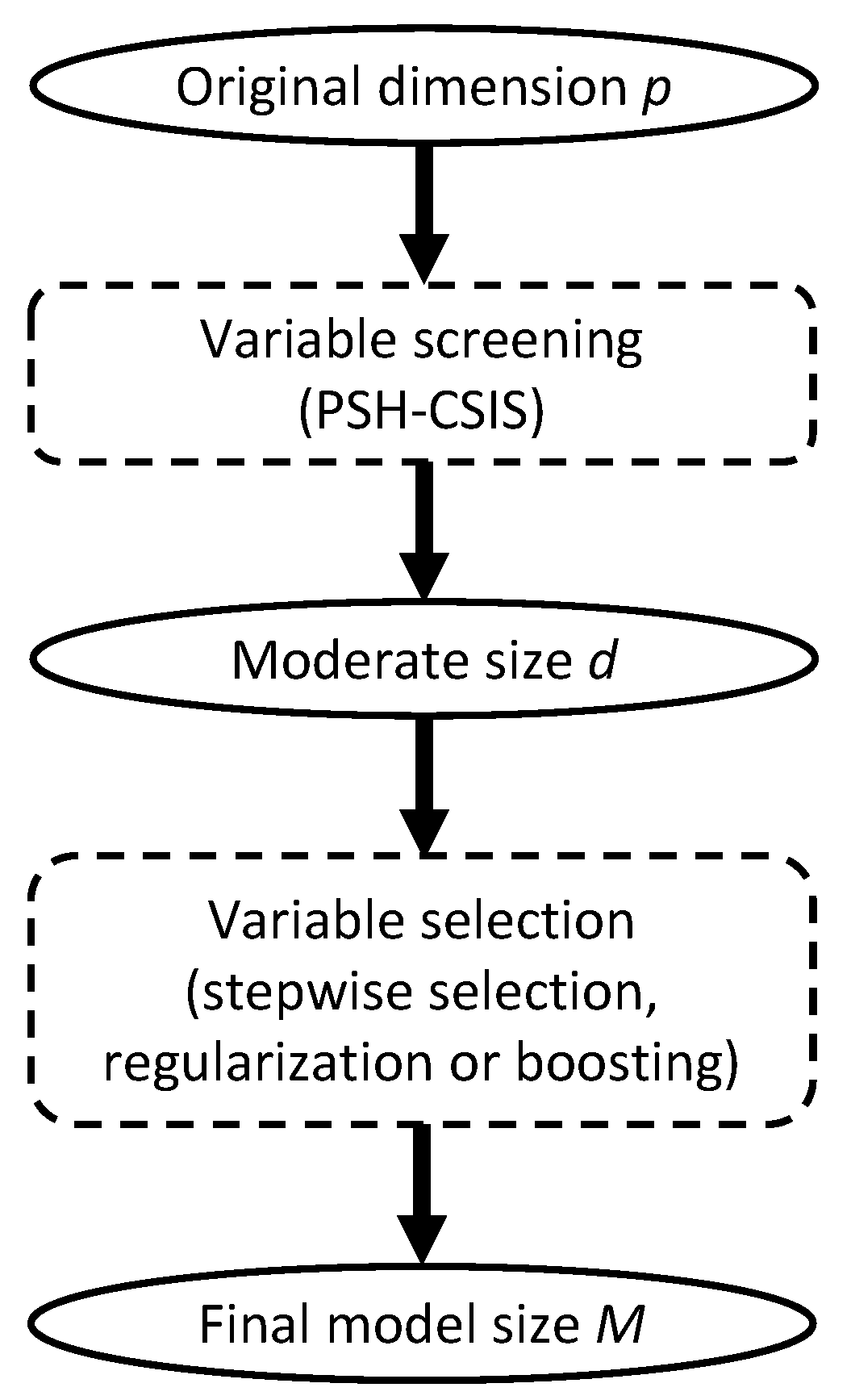
2. Materials and Methods
2.1. The Proportional Subdistribution Hazards Model
2.2. Conditional Sure Independence Screening for PSH
2.3. Non-Muscle-Invasive Bladder Carcinoma Data
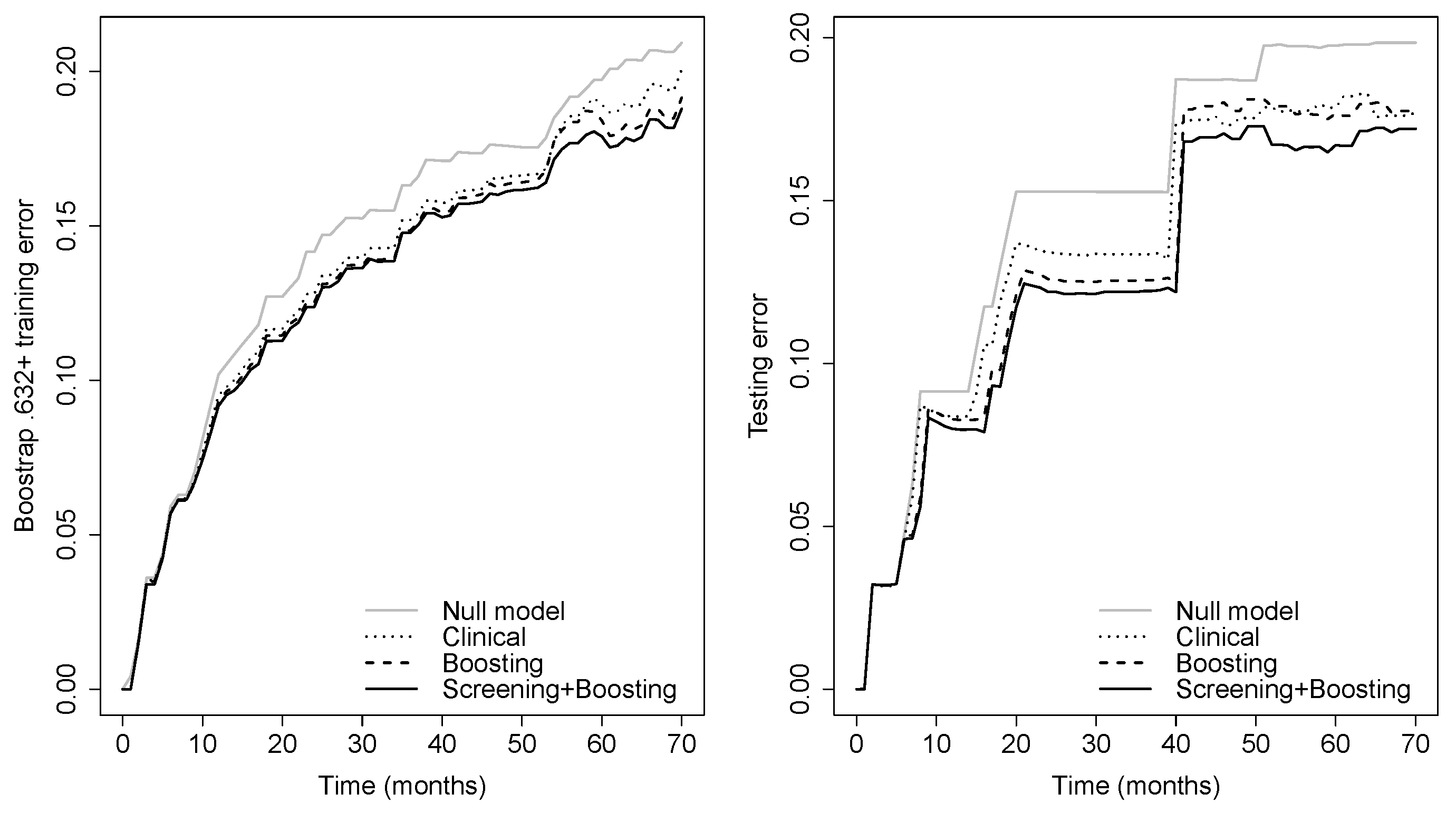
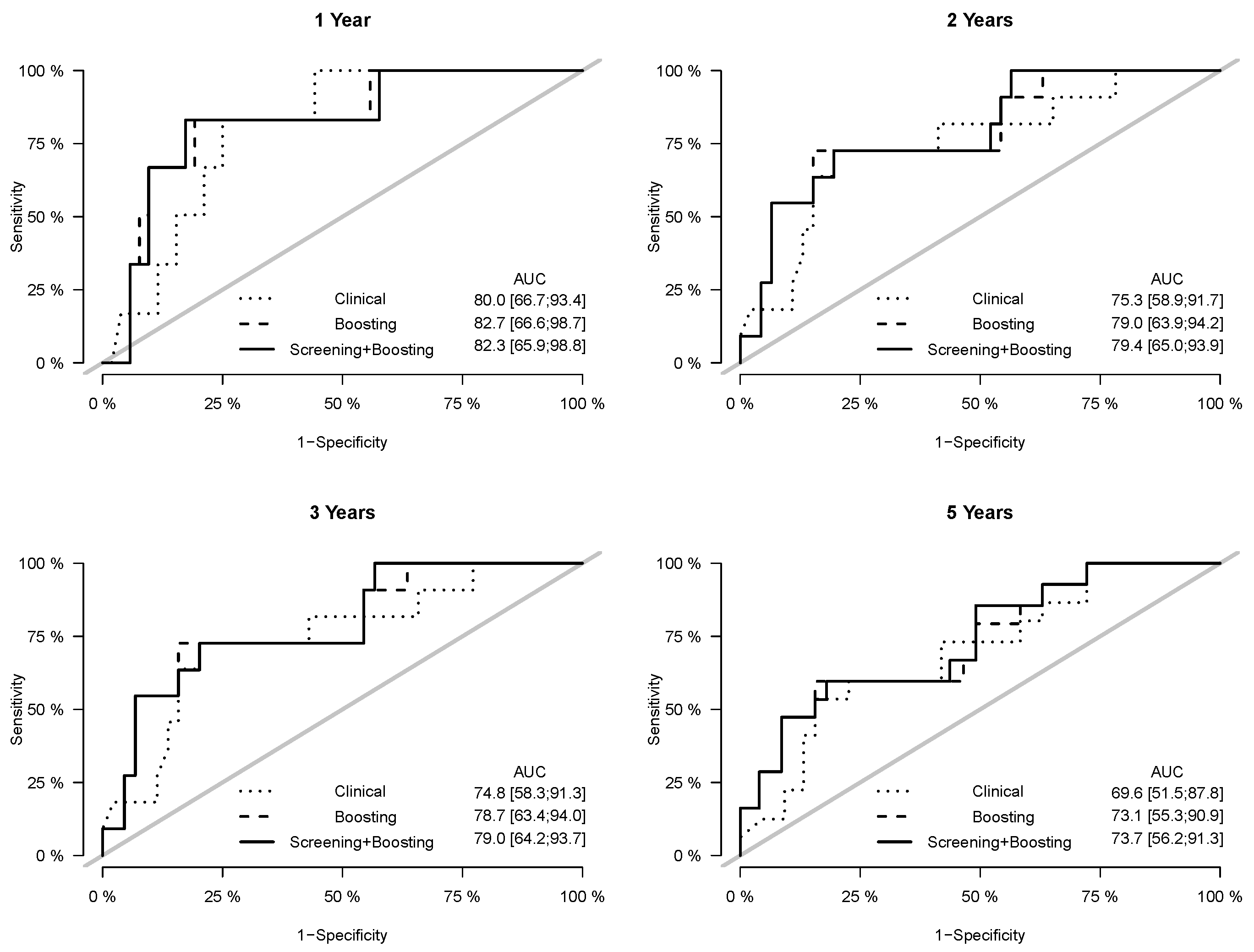
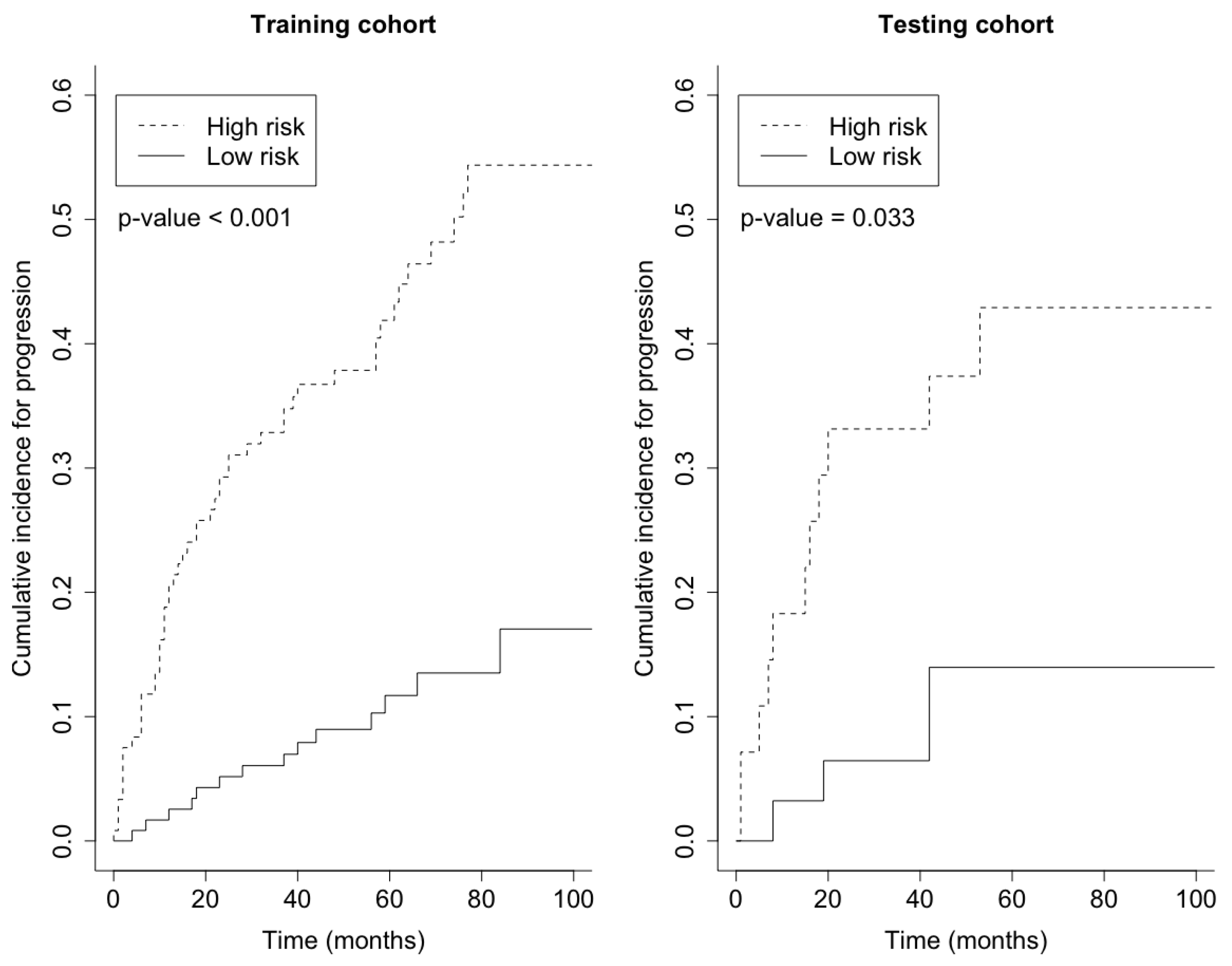
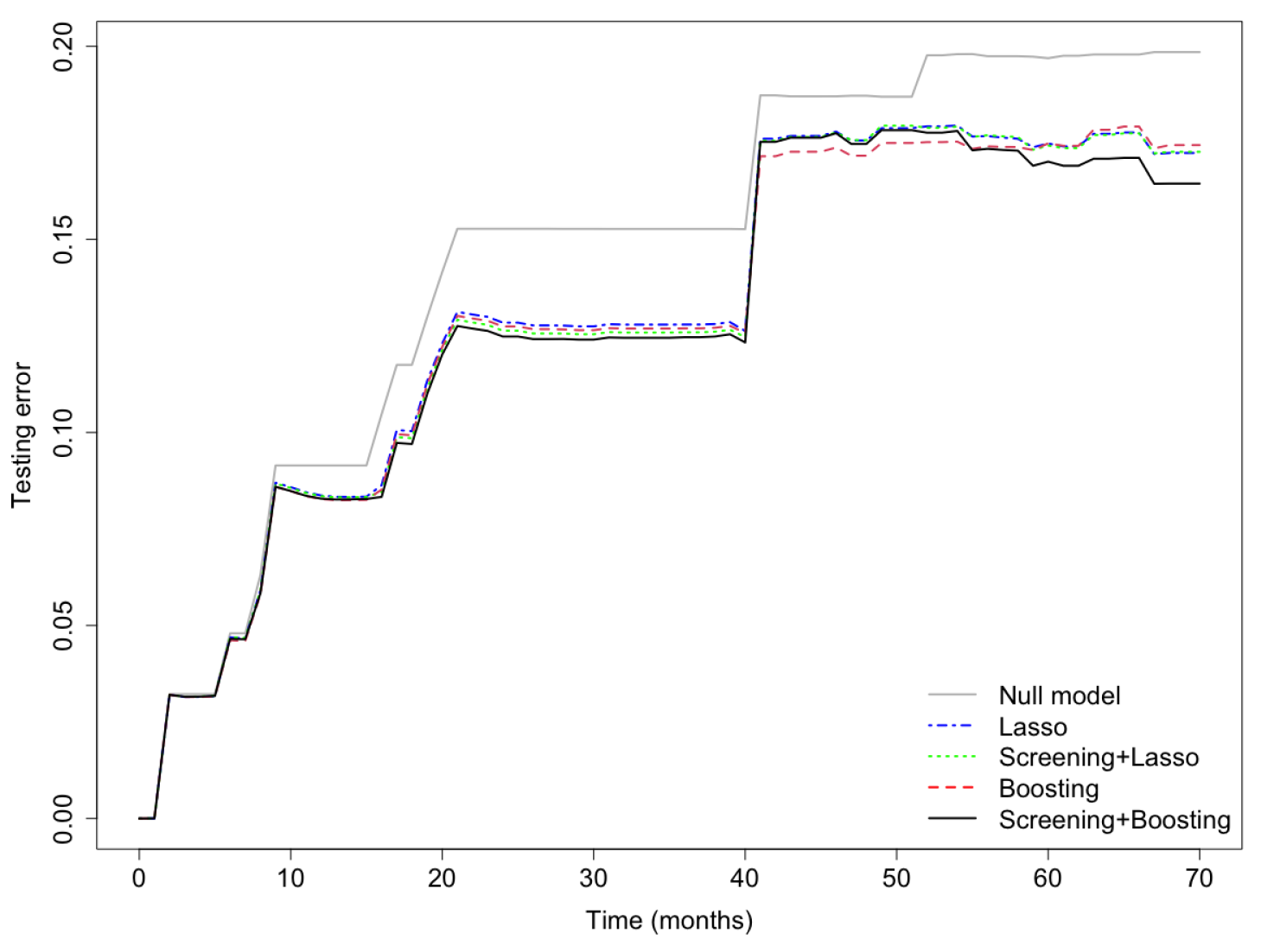
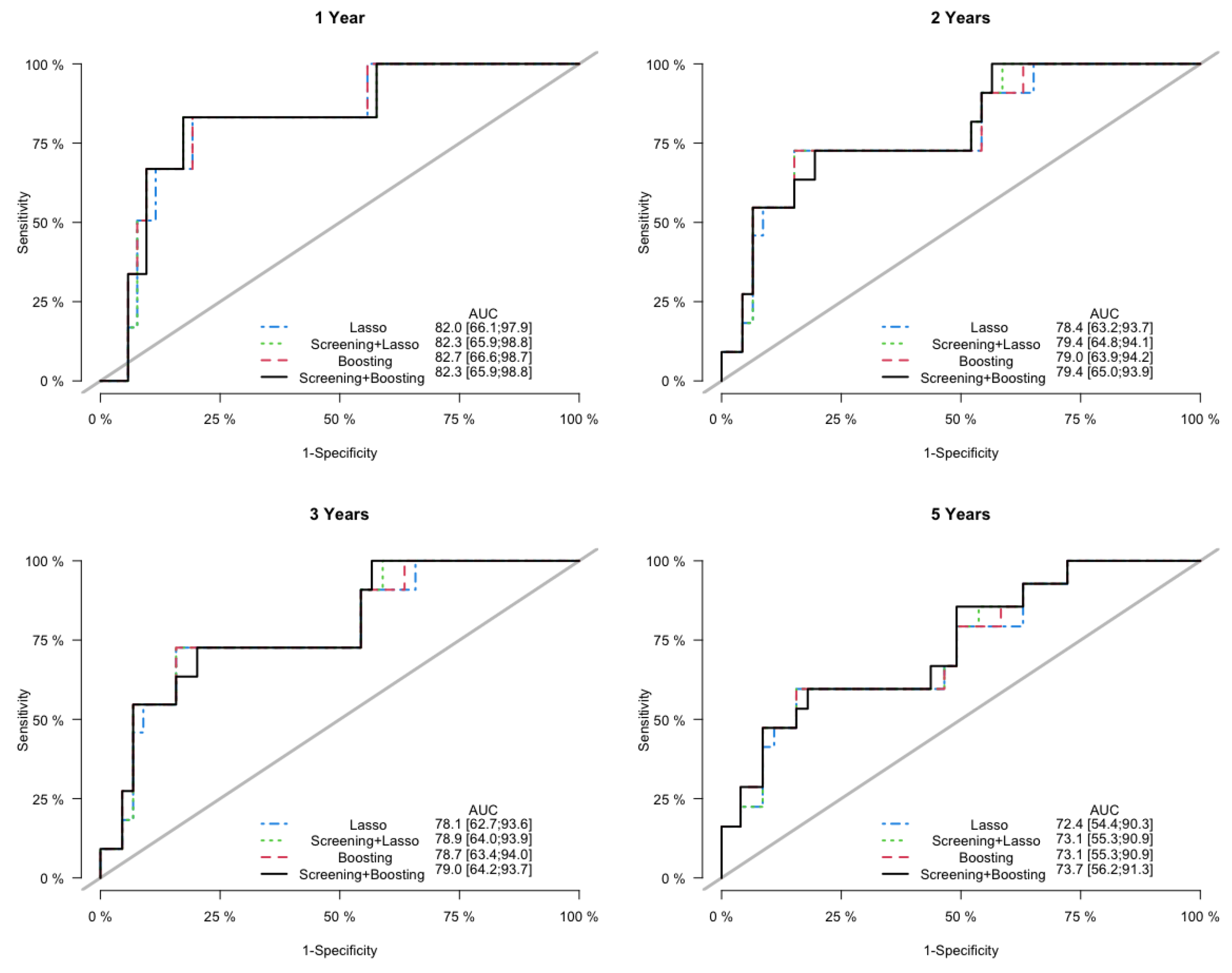
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teoh, J.Y.C.; Huang, J.; Ko, W.Y.K.; Lok, V.; Choi, P.; Ng, C.F.; Sengupta, S.; Mostafid, H.; Kamat, A.M.; Black, P.C.; et al. Global trends of bladder cancer incidence and mortality, and their associations with tobacco use and gross domestic product per capita. Eur. Urol. 2020, 78, 893–906. [Google Scholar] [CrossRef]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Cookson, M.S.; Herr, H.W.; Zhang, Z.F.; Soloway, S.; Sogani, P.C.; Fair, W.R. The treated natural history of high risk superficial bladder cancer: 15-year outcome. J. Urol. 1997, 158, 62–67. [Google Scholar] [CrossRef]
- Dignam, J.J.; Zhang, Q.; Kocherginsky, M. The Use and Interpretation of Competing Risks Regression ModelsModeling with Competing Risks. Clin. Cancer Res. 2012, 18, 2301–2308. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.P.; Gray, R.J. A proportional hazards model for the subdistribution of a competing risk. J. Am. Stat. Assoc. 1999, 94, 496–509. [Google Scholar] [CrossRef]
- Fu, Z.; Parikh, C.R.; Zhou, B. Penalized variable selection in competing risks regression. Lifetime Data Anal. 2017, 23, 353–376. [Google Scholar] [CrossRef]
- Hou, J.; Bradic, J.; Xu, R. Inference under fine-gray competing risks model with high-dimensional covariates. Electron. J. Stat. 2019, 13, 4449–4507. [Google Scholar] [CrossRef]
- Kawaguchi, E.S.; Shen, J.I.; Suchard, M.A.; Li, G. Scalable algorithms for large competing risks data. J. Comput. Graph. Stat. 2021, 30, 685–693. [Google Scholar] [CrossRef]
- Tapak, L.; Kosorok, M.R.; Sadeghifar, M.; Hamidi, O.; Afshar, S.; Doosti, H. Regularized Weighted Nonparametric Likelihood Approach for High-Dimension Sparse Subdistribution Hazards Model for Competing Risk Data. Comput. Math. Methods Med. 2021, 2021, 5169052. [Google Scholar] [CrossRef]
- Sun, H.; Wang, X. High-dimensional feature selection in competing risks modeling: A stable approach using a split-and-merge ensemble algorithm. Biom. J. 2022. [Google Scholar] [CrossRef]
- Bühlmann, P.; Yu, B. Boosting with the L 2 loss: Regression and classification. J. Am. Stat. Assoc. 2003, 98, 324–339. [Google Scholar] [CrossRef]
- Binder, H.; Allignol, A.; Schumacher, M.; Beyersmann, J. Boosting for high-dimensional time-to-event data with competing risks. Bioinformatics 2009, 25, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Lv, J. Sure independence screening for ultrahigh dimensional feature space. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2008, 70, 849–911. [Google Scholar] [CrossRef] [Green Version]
- Barut, E.; Fan, J.; Verhasselt, A. Conditional sure independence screening. J. Am. Stat. Assoc. 2016, 111, 1266–1277. [Google Scholar] [CrossRef]
- Hong, H.G.; Li, Y. Feature selection of ultrahigh-dimensional covariates with survival outcomes: A selective review. Appl. Math.-A J. Chin. Univ. 2017, 32, 379–396. [Google Scholar] [CrossRef] [Green Version]
- Kuk, D.; Varadhan, R. Model selection in competing risks regression. Stat. Med. 2013, 32, 3077–3088. [Google Scholar] [CrossRef]
- Gray, R.J. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann. Stat. 1988, 16, 1141–1154. [Google Scholar] [CrossRef]
- Li, R.; Zhong, W.; Zhu, L. Feature screening via distance correlation learning. J. Am. Stat. Assoc. 2012, 107, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyrskjøt, L.; Zieger, K.; Real, F.X.; Malats, N.; Carrato, A.; Hurst, C.; Kotwal, S.; Knowles, M.; Malmström, P.U.; de la Torre, M.; et al. Gene expression signatures predict outcome in non–muscle-invasive bladder carcinoma: A multicenter validation study. Clin. Cancer Res. 2007, 13, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Fölsch, H.; Ohno, H.; Bonifacino, J.S.; Mellman, I. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell 1999, 99, 189–198. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Cai, Y.; Changyong, E.; Sheng, J. Screening and validation of independent predictors of poor survival in pancreatic cancer. Pathol. Oncol. Res. 2021, 27, 1609868. [Google Scholar]
- Wu, H.; Fan, L.; Liu, H.; Guan, B.; Hu, B.; Liu, F.; Hocher, B.; Yin, L. Identification of key genes and prognostic analysis between chromophobe renal cell carcinoma and renal oncocytoma by bioinformatic analysis. BioMed Res. Int. 2020, 2020, 4030915. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, Q.; Shen, Y.; Gao, Y.; Fan, X.; Chen, S.; Ye, X.; Xu, J. System analysis of adaptor-related protein complex 1 subunit mu 2 (AP1M2) on malignant tumors: A pan-cancer analysis. J. Oncol. 2022, 2022, 7945077. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.O.; Bacchetti, T.; Ferretti, G. Alterations of antioxidant enzymes and biomarkers of nitro-oxidative stress in tissues of bladder cancer. Oxidative Med. Cell. Longev. 2019, 2019, 2730896. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, E.; Jablonowski, Z.; Tomasik, B.; Gromadzinska, J.; Jablonska, E.; Konecki, T.; Fendler, W.; Sosnowski, M.; Wasowicz, W.; Reszka, E. Different gene expression and activity pattern of antioxidant enzymes in bladder cancer. Anticancer Res. 2017, 37, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Keil, R.; Schulz, J.; Hatzfeld, M. p0071/PKP4, a multifunctional protein coordinating cell adhesion with cytoskeletal organization. Biol. Chem. 2013, 394, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Setzer, S.V.; Calkins, C.C.; Garner, J.; Summers, S.; Green, K.J.; Kowalczyk, A.P. Comparative analysis of armadillo family proteins in the regulation of a431 epithelial cell junction assembly, adhesion and migration. J. Investig. Dermatol. 2004, 123, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Nakatsuji, H.; Takahashi, M.; Avirmed, S.; Fukawa, T.; Takemura, M.; Fukumori, T.; Kanayama, H. Up-regulation of plakophilin-2 and Down-regulation of plakophilin-3 are correlated with invasiveness in bladder cancer. Urology 2012, 79, 240.e1–240.e8. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S. Chronic inflammation and bladder cancer. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2007; Volume 25, pp. 260–268. [Google Scholar]
- Ntanasis-Stathopoulos, I.; Fotiou, D.; Terpos, E. CCL3 signaling in the tumor microenvironment. Tumor Microenviron. 2020, 1231, 13–21. [Google Scholar]
- Eruslanov, E.; Neuberger, M.; Daurkin, I.; Perrin, G.Q.; Algood, C.; Dahm, P.; Rosser, C.; Vieweg, J.; Gilbert, S.M.; Kusmartsev, S. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int. J. Cancer 2012, 130, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Wang, G.; Shi, Y.; Xu, H.; Zheng, Y.; Chen, Y. MCMs in cancer: Prognostic potential and mechanisms. Anal. Cell. Pathol. 2020, 2020, 3750294. [Google Scholar] [CrossRef]
- Fristrup, N.; Birkenkamp-Demtröder, K.; Reinert, T.; Sanchez-Carbayo, M.; Segersten, U.; Malmström, P.U.; Palou, J.; Alvarez-Múgica, M.; Pan, C.C.; Ulhøi, B.P.; et al. Multicenter validation of Cyclin D1, MCM7, TRIM29, and UBE2C as prognostic protein markers in non-muscle–invasive bladder cancer. Am. J. Pathol. 2013, 182, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigehara, K.; Sasagawa, T.; Kawaguchi, S.; Nakashima, T.; Shimamura, M.; Maeda, Y.; Konaka, H.; Mizokami, A.; Koh, E.; Namiki, M. Etiologic role of human papillomavirus infection in bladder carcinoma. Cancer 2011, 117, 2067–2076. [Google Scholar] [CrossRef]
- Zhang, J.X.; Chen, Z.H.; Chen, D.L.; Tian, X.P.; Wang, C.Y.; Zhou, Z.W.; Gao, Y.; Xu, Y.; Chen, C.; Zheng, Z.S.; et al. LINC01410-miR-532-NCF2-NF-kB feedback loop promotes gastric cancer angiogenesis and metastasis. Oncogene 2018, 37, 2660–2675. [Google Scholar] [CrossRef] [Green Version]
- Muthuswamy, R.; Wang, L.; Pitteroff, J.; Gingrich, J.R.; Kalinski, P. Combination of IFNα and poly-I: C reprograms bladder cancer microenvironment for enhanced CTL attraction. J. Immunother. Cancer 2015, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhijn, B.W.; Burger, M.; Lotan, Y.; Solsona, E.; Stief, C.G.; Sylvester, R.J.; Witjes, J.A.; Zlotta, A.R. Recurrence and progression of disease in non–muscle-invasive bladder cancer: From epidemiology to treatment strategy. Eur. Urol. 2009, 56, 430–442. [Google Scholar] [CrossRef]
- van Rhijn, B.W. Combining molecular and pathologic data to prognosticate non-muscle-invasive bladder cancer. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2012; Volume 30, pp. 518–523. [Google Scholar]
- di Meo, N.A.; Loizzo, D.; Pandolfo, S.D.; Autorino, R.; Ferro, M.; Porta, C.; Stella, A.; Bizzoca, C.; Vincenti, L.; Crocetto, F.; et al. Metabolomic Approaches for Detection and Identification of Biomarkers and Altered Pathways in Bladder Cancer. Int. J. Mol. Sci. 2022, 23, 4173. [Google Scholar] [CrossRef]
- Bellach, A.; Kosorok, M.R.; Rüschendorf, L.; Fine, J.P. Weighted NPMLE for the subdistribution of a competing risk. J. Am. Stat. Assoc. 2019, 114, 259–270. [Google Scholar] [CrossRef]
- Tian, B.; Liu, Z.; Wang, H. Non-marginal feature screening for varying coefficient competing risks model. Stat. Probab. Lett. 2022, 190, 109648. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Frequency (Percent) |
|---|---|
| Age | |
| Less than 60 | 42 (14.0%) |
| 60–69 | 67 (24.0%) |
| 70–79 | 105 (35.0%) |
| 80 or greater | 81 (27.0%) |
| Gender | |
| Female | 59 (19.7%) |
| Male | 241 (80.3%) |
| WHO Grade | |
| High | 176 (58.7%) |
| Low | 124 (41.3%) |
| Stage | |
| Ta | 173 (57.7%) |
| T1 | 127 (42.3%) |
| Treatment | |
| BCG/MMC | 82 (27.3%) |
| None | 218 (72.7%) |
| Model | Gene Selected |
|---|---|
| PSH-CSIS + CoxBoost | AP1M2(-), CAT(-), CCL3(+), MCM7(+), NCF2(+), PKP4(-) |
| CoxBoost | AP1M2(-), CAT(-), CCL3(+), MCM7(+), NCF2(+) |
| Variable | Univariate Analysis | Multivariable Analysis | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | p-Value | Hazard Ratio (95% CI) | p-Value | |
| 6-gene signature | 8.95 (4.75, 16.90) | <0.001 | 12.55 (6.11, 25.80) | <0.001 |
| Age | ||||
| Age > 70 | 1.70 (1.11, 2.60) | 0.015 | 1.55 (0.99, 2.45) | 0.058 |
| Age ≤ 70 | - | - | - | - |
| Gender | ||||
| Male | 0.80 (0.43, 1.33) | 0.38 | 0.84 (0.46, 1.55) | 0.580 |
| Female | - | - | - | - |
| WHO Grade | ||||
| low | 0.40 (0.28, 0.64) | <0.001 | 0.43 (0.24, 0.77) | 0.005 |
| high | - | - | - | - |
| Stage | ||||
| Ta | 0.63 (0.41, 0.97) | 0.034 | 1.66 (0.99, 2.80) | 0.056 |
| T2 | - | - | - | - |
| Treatment | ||||
| None | 2.04 (1.19, 3.48) | 0.009 | 2.60 (1.44, 4.69) | 0.002 |
| BCG/MMC | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, C.; Bandyopadhyay, D.; Sarkar, D. Gene Screening for Prognosis of Non-Muscle-Invasive Bladder Carcinoma under Competing Risks Endpoints. Cancers 2023, 15, 379. https://doi.org/10.3390/cancers15020379
Ke C, Bandyopadhyay D, Sarkar D. Gene Screening for Prognosis of Non-Muscle-Invasive Bladder Carcinoma under Competing Risks Endpoints. Cancers. 2023; 15(2):379. https://doi.org/10.3390/cancers15020379
Chicago/Turabian StyleKe, Chenlu, Dipankar Bandyopadhyay, and Devanand Sarkar. 2023. "Gene Screening for Prognosis of Non-Muscle-Invasive Bladder Carcinoma under Competing Risks Endpoints" Cancers 15, no. 2: 379. https://doi.org/10.3390/cancers15020379
APA StyleKe, C., Bandyopadhyay, D., & Sarkar, D. (2023). Gene Screening for Prognosis of Non-Muscle-Invasive Bladder Carcinoma under Competing Risks Endpoints. Cancers, 15(2), 379. https://doi.org/10.3390/cancers15020379