
Tissue-Specific microRNA Expression Profiling to Derive Novel Biomarkers for the Diagnosis and Subtyping of Small B-Cell Lymphomas

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. RNA Isolation, Reverse-Transcription, cDNA Amplification, and Real-Time qPCR
2.3. Data Processing and Statistical Analysis
2.4. Subtype Classification and Machine Learning
2.5. Pathway Analysis of Differentially Expressed miRNAs
3. Results
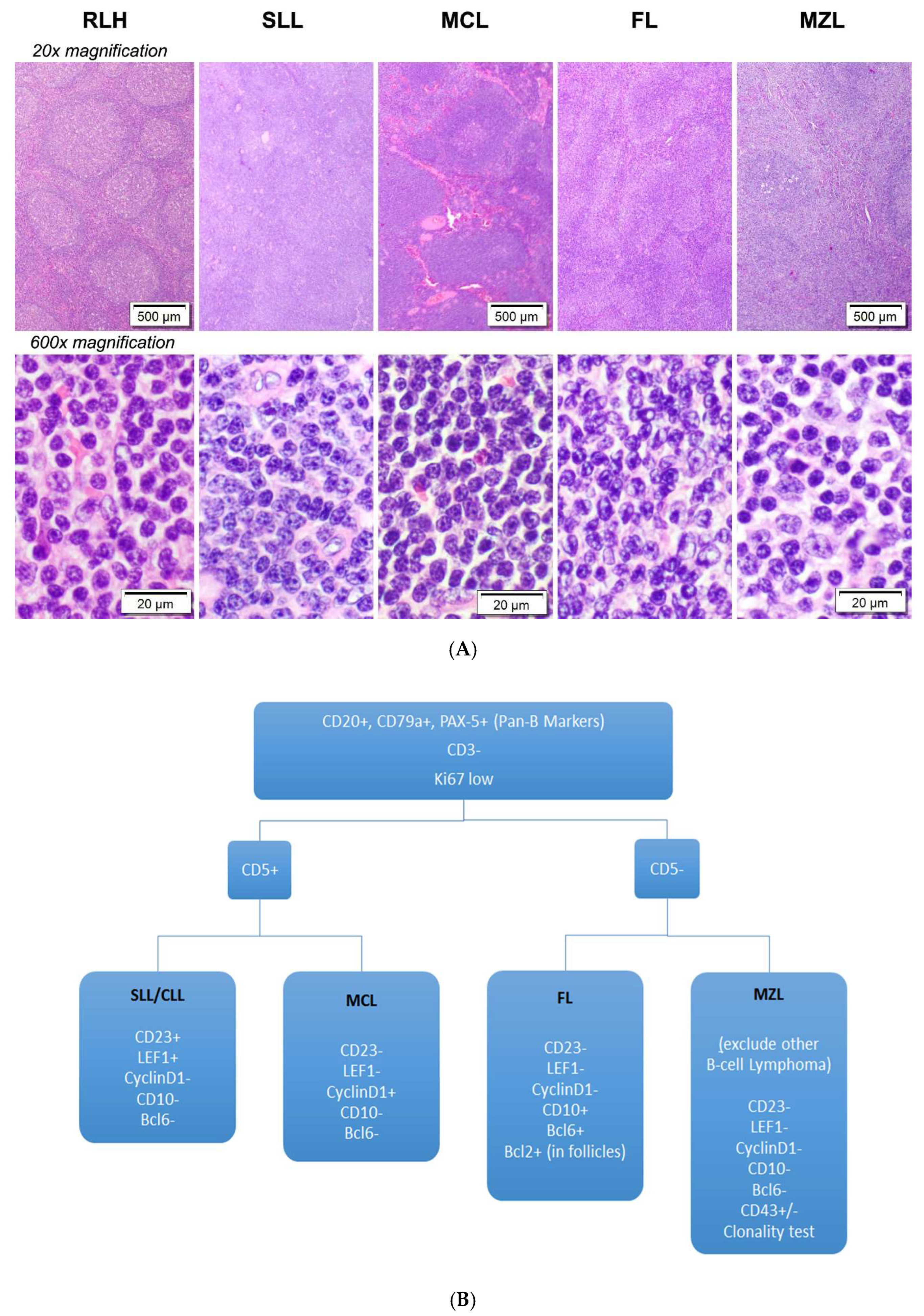
3.1. Diagnostic Challenges of Small B-Cell Lymphomas
3.2. Consistency of miRNAs Expression across FFPE Samples in the Discovery and Validation Cohorts
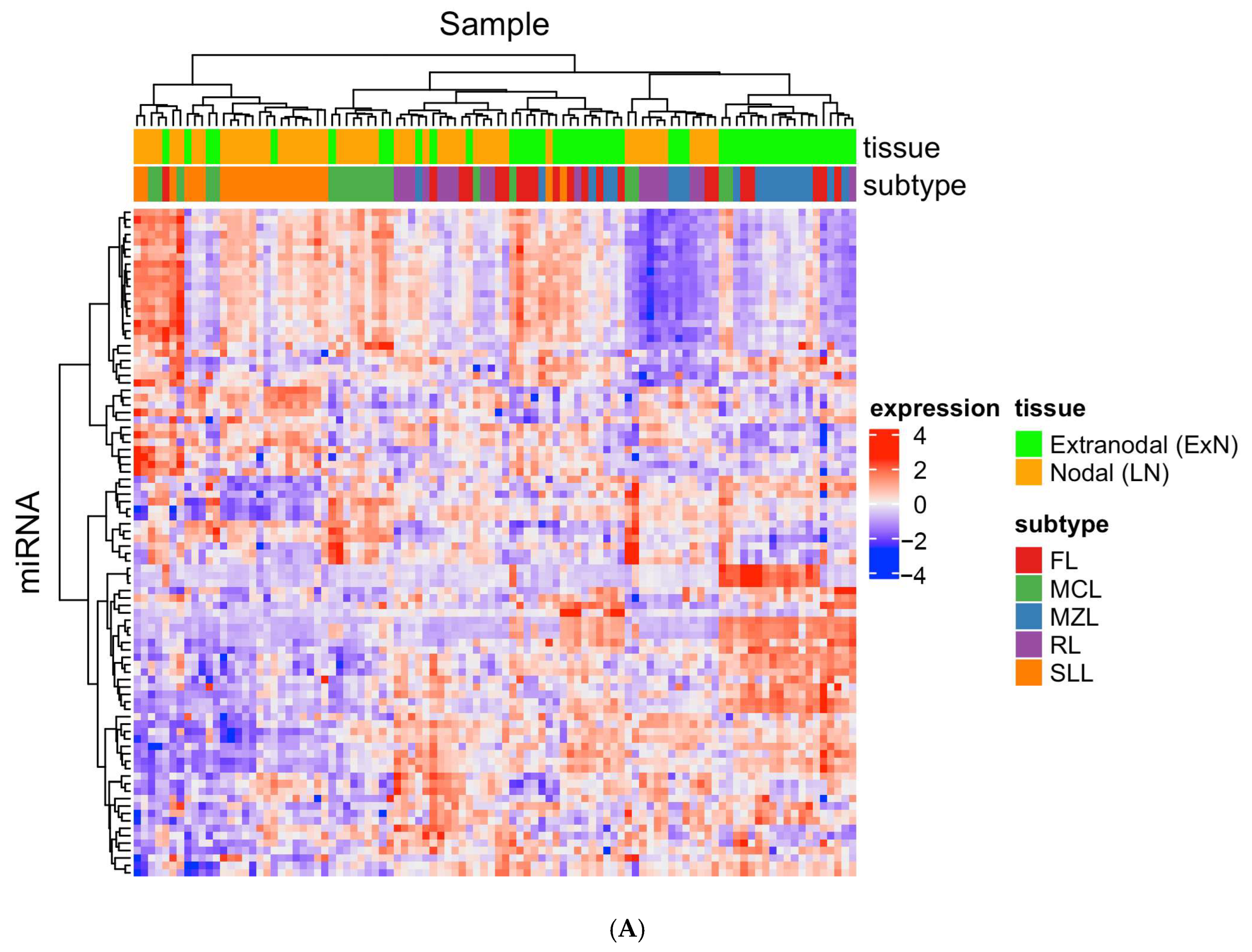
3.3. miRNAs Signature can Differentiate Lymphoma from Reactive Lymphoid Proliferation
3.4. miRNAs Signature can Subtype Small B-Cell Lymphomas
3.5. miRNA Expression could Infer Meaningful Biological Differences between Reactive and Neoplastic Lymphoid Proliferation
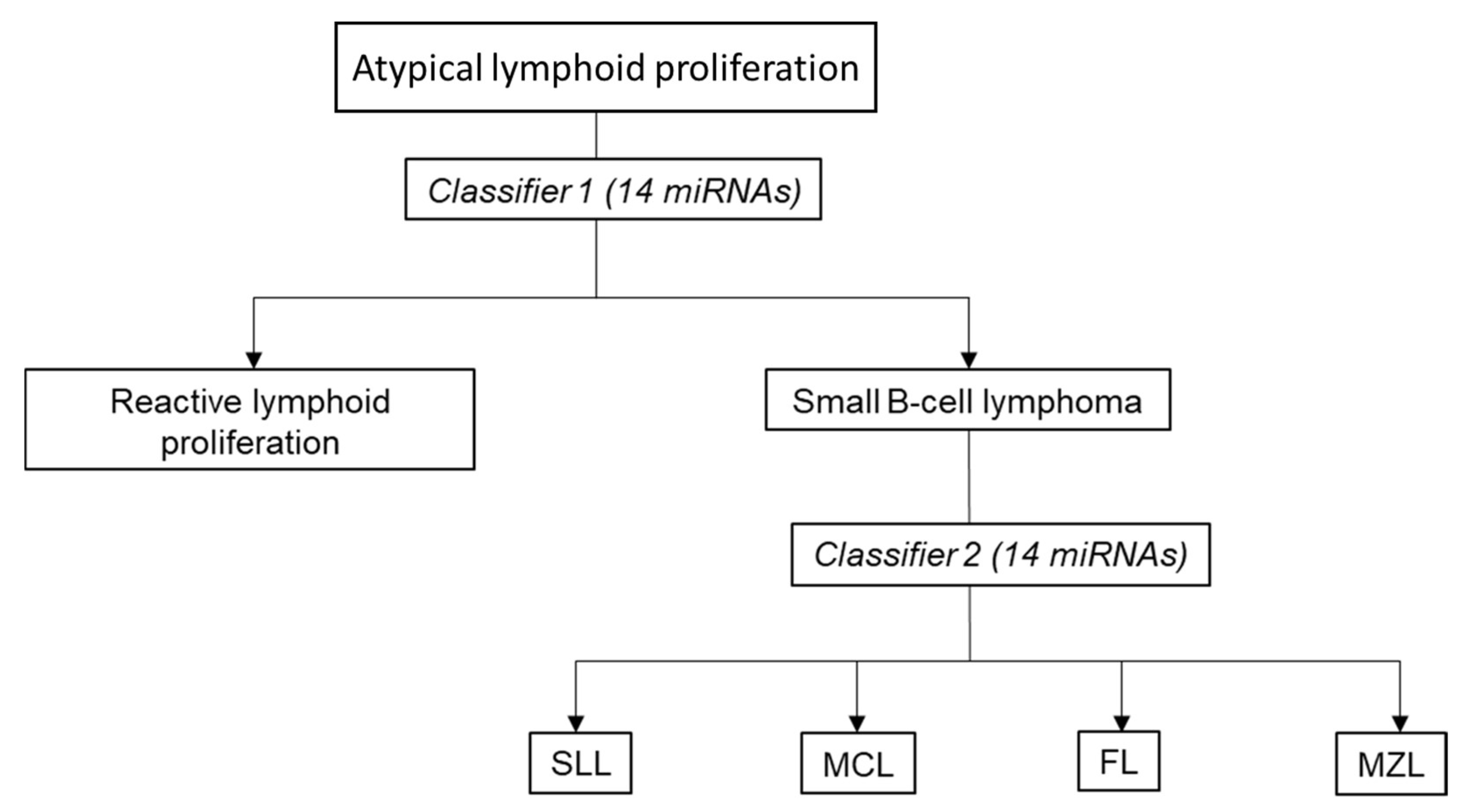
3.6. Proposed Two-Stage Diagnostic Algorithm for miRNA-Based Classification of Small B-Cell Lymphomas
4. Discussion
4.1. miRNAs as Potential Diagnostic Biomarkers
4.2. Biological Relevance of miRNA Biomarkers
4.3. Possible Pathways Implicated by miRNAs
4.3.1. Cytosolic DNA Sensing Pathway, RIG-I-like Receptor Signaling Pathway and NOD-like Receptor Signaling Pathway
4.3.2. Gɑ12/13 Signaling Events
4.3.3. Other Notable Signaling Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, J.K.; Kwong, Y.-L. Common misdiagnoses in lymphomas and avoidance strategies. Lancet Oncol. 2010, 11, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Zu, Y. Diagnostic Algorithm of Common Mature B-Cell Lymphomas by Immunohistochemistry. Arch. Pathol. Lab. Med. 2017, 141, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harfe, B.D. MicroRNAs in vertebrate development. Curr. Opin. Genet. Dev. 2005, 15, 410–415. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Wan, G.; En Lim, Q.; Too, H.-P. High-performance quantification of mature microRNAs by real-time RT-PCR using deoxyuridine-incorporated oligonucleotides and hemi-nested primers. RNA 2010, 16, 1436–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, R.; Loke, S.Y.; Tan, V.K.-M.; Quek, S.T.; Jagmohan, P.; Tang, Y.C.; Madhukumar, P.; Tan, B.K.-T.; Yong, W.S.; Sim, Y.; et al. Development of a microRNA Panel for Classification of Abnormal Mammograms for Breast Cancer. Cancers 2021, 13, 2130. [Google Scholar] [CrossRef]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Mestdagh, P.; Feys, T.; Bernard, N.; Guenther, S.; Chen, C.; Speleman, F.; Vandesompele, J. High-throughput stem-loop RT-qPCR miRNA expression profiling using minute amounts of input RNA. Nucleic Acids Res. 2008, 36, 143. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostat. Oxf. Engl. 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien. R Package Version 1.7-11. 2022. Available online: https://CRAN.R-project.org/package=e1071 (accessed on 10 November 2022).
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Fernandez-Mercado, M.; Manterola, L.; Lawrie, C.H. MicroRNAs in Lymphoma: Regulatory Role and Biomarker Potential. Curr. Genom. 2015, 16, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Liu, C.G.; Sevignani, C.; Ferracin, M.; Felli, N.; Dumitru, C.D.; Shimizu, M.; Cimmino, A.; Zupo, S.; Dono, M.; et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. USA 2004, 101, 11755–11760. [Google Scholar] [CrossRef] [Green Version]
- Lawrie, C.H. MicroRNAs and lymphomagenesis: A functional review. Br. J. Haematol. 2013, 160, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Johanson, T.M.; Skinner, J.P.J.; Kumar, A.; Zhan, Y.; Lew, A.M.; Chong, M.M.W. The role of microRNAs in lymphopoiesis. Int. J. Hematol. 2014, 100, 246–253. [Google Scholar] [CrossRef]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.M.; Cheng, H.; Tang, Y.C.; Leong, S.M.; Teo, P.Y.; Lee, C.K.; Lee, V.K.M.; Hue, S.S. MicroRNAs as Potential Biomarkers in the Differential Diagnosis of Lipomatous Tumors and Their Mimics. Int. J. Mol. Sci. 2022, 23, 7804. [Google Scholar] [CrossRef]
- Gilad, S.; Lithwick-Yanai, G.; Barshack, I.; Benjamin, S.; Krivitsky, I.; Edmonston, T.B.; Bibbo, M.; Thurm, C.; Horowitz, L.; Huang, Y.; et al. Classification of the Four Main Types of Lung Cancer Using a MicroRNA-Based Diagnostic Assay. J. Mol. Diagn. 2012, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Fridman, E.; Dotan, Z.; Barshack, I.; David, M.B.; Dov, A.; Tabak, S.; Zion, O.; Benjamin, S.; Benjamin, H.; Kuker, H.; et al. Accurate Molecular Classification of Renal Tumors Using MicroRNA Expression. J. Mol. Diagn. 2010, 12, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, H.; Lebanony, D.; Rosenwald, S.; Cohen, L.; Gibori, H.; Barabash, N.; Ashkenazi, K.; Goren, E.; Meiri, E.; Morgenstern, S.; et al. A Diagnostic Assay Based on MicroRNA Expression Accurately Identifies Malignant Pleural Mesothelioma. J. Mol. Diagn. 2010, 12, 771–779. [Google Scholar] [CrossRef]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced Expression of the let-7 MicroRNAs in Human Lung Cancers in Association with Shortened Postoperative Survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef]
- Kowalik, C.G.; Palmer, D.A.; Sullivan, T.B.; Teebagy, P.A.; Dugan, J.M.; Libertino, J.A.; Burks, E.J.; Canes, D.; Rieger-Christ, K.M. Profiling microRNA from nephrectomy and biopsy specimens: Predictors of progression and survival in clear cell renal cell carcinoma. BJU Int. 2017, 120, 428–440. [Google Scholar] [CrossRef] [Green Version]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, S.T.; Chan, T.L.; Kwong, D.L.; Au, G.K.; et al. Microrna expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Scholl, V.; Hassan, R.; Zalcberg, I.R. miRNA-451: A putative predictor marker of Imatinib therapy response in chronic myeloid leukemia. Leuk. Res. 2012, 36, 119–121. [Google Scholar] [CrossRef]
- Vecchione, A.; Belletti, B.; Lovat, F.; Volinia, S.; Chiappetta, G.; Giglio, S.; Sonego, M.; Cirombella, R.; Onesti, E.C.; Pellegrini, P.; et al. A microRNA signature defines chemoresistance in ovarian cancer through modulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9845–9850. [Google Scholar] [CrossRef]
- Ji, J.; Shi, J.; Budhu, A.; Yu, Z.; Forgues, M.; Roessler, S.; Ambs, S.; Chen, Y.; Meltzer, P.S.; Croce, C.M.; et al. MicroRNA Expression, Survival, and Response to Interferon in Liver Cancer. N. Engl. J. Med. 2009, 361, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Bovell, L.; Shanmugam, C.; Katkoori, V.R.; Zhang, B.; Vogtmann, E.; Grizzle, W.E.; Manne, U. microRNAs are Stable in Formalin-Fixed Paraffin-Embedded Archival Tissue Specimens of Colorectal Cancers Stored for up to 28 Years. Front. Biosci. Elite Ed. 2012, 4, 1937–1940. [Google Scholar]
- Doleshal, M.; Magotra, A.A.; Choudhury, B.; Cannon, B.D.; Labourier, E.; Szafranska, A.E. Evaluation and Validation of Total RNA Extraction Methods for MicroRNA Expression Analyses in Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn. 2008, 10, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, N.; Aharonov, R.; Meiri, E.; Rosenwald, S.; Spector, Y.; Zepeniuk, M.; Benjamin, H.; Shabes, N.; Tabak, S.; Levy, A.; et al. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 2008, 26, 462–469. [Google Scholar] [CrossRef]
- Szafranska, A.E.; Davison, T.S.; Shingara, J.; Doleshal, M.; Riggenbach, J.A.; Morrison, C.D.; Jewell, S.; Labourier, E. Accurate Molecular Characterization of Formalin-Fixed, Paraffin-Embedded Tissues by microRNA Expression Profiling. J. Mol. Diagn. 2008, 10, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ruan, K. MicroRNA detection by microarray. Anal. Bioanal. Chem. 2009, 394, 1117–1124. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Lee, E.J.; Jiang, J.; Sarkar, A.; Yang, L.; Elton, T.S.; Chen, C. Real-time PCR quantification of precursor and mature microRNA. Methods 2008, 44, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 2005, 33, 179. [Google Scholar] [CrossRef]
- Murphy, J.B.S. Reliability of real-time reverse-transcription PCR in clinical diagnostics: Gold standard or substandard? Expert Rev. Mol. Diagn. 2009, 9, 187–197. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of Myc-Mediated miR-29 by HDAC3 and EZH2 As a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef]
- Zhang, J.; Jima, D.D.; Jacobs, C.; Fischer, R.; Gottwein, E.; Huang, G.; Lugar, P.L.; Lagoo, A.S.; Rizzieri, D.A.; Friedman, D.R.; et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood 2009, 113, 4586–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Zhang, Q.; Wu, S.; Yan, S.; Zhao, R.; Sun, Y.; Tian, X.; Zhou, K. MiRNA-223-3p Affects Mantle Cell Lymphoma Development by Regulating the CHUK/NF-ƘB2 Signaling Pathway. OncoTargets Ther. 2021, 14, 1553–1564. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Lai, H.F.; Huang, T.C.; Chen, Y.G.; Ye, R.H.; Chang, P.Y.; Lai, S.W.; Chen, Y.C.; Lee, C.H.; Liu, W.N.; et al. Aberrantly reduced expression of miR-342-5p contributes to CCND1-associated chronic myeloid leukemia progression and imatinib resistance. Cell Death Dis. 2021, 12, 908. [Google Scholar] [CrossRef]
- Vose, J.M. Mantle cell lymphoma: 2017 update on diagnosis, risk-stratification, and clinical management. Am. J. Hematol. 2017, 92, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmrich, S.; Engeland, F.; El-Khatib, M.; Henke, K.; Obulkasim, A.; Schöning, J.; Katsman-Kuipers, J.E.; Michel Zwaan, C.; Pich, A.; Stary, J.; et al. miR-139-5p controls translation in myeloid leukemia through EIF4G2. Oncogene 2016, 35, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Demosthenous, C.; Han, J.J.; Stenson, M.J.; Maurer, M.J.; Wellik, L.E.; Link, B.; Hege, K.; Dogan, A.; Sotomayor, E.; Witzig, T.; et al. Translation initiation complex eIF4F is a therapeutic target for dual mTOR kinase inhibitors in non-Hodgkin lymphoma. Oncotarget 2015, 6, 9488–9501. [Google Scholar] [CrossRef] [Green Version]
- Lone, W.; Bouska, A.; Sharma, S.; Amador, C.; Saumyaranjan, M.; Herek, T.A.; Heavican, T.B.; Yu, J.; Lim, S.T.; Ong, C.K.; et al. Genome-wide microRNA expression profiling of molecular subgroups of peripheral T-cell lymphoma. Clin. Cancer Res. 2021, 27, 6039–6053. [Google Scholar] [CrossRef]
- Gao, Y.; Ding, X. miR-145-5p exerts anti-tumor effects in diffuse large B-cell lymphoma by regulating S1PR1/STAT3/AKT pathway. Leuk. Lymphoma. 2021, 62, 1884–1891. [Google Scholar] [CrossRef]
- Lin, D.; Wang, Y.; Lei, L.; Lin, C. Circ_0003645 serves as miR-335-5p sponge to promote the biological process of diffuse large B-cell lymphoma by upregulating NFIB. Autoimmunity 2022, 55, 127–135. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Tao, Y.; Chen, H.; Qin, Y.; He, X.; Zhou, S.; Liu, P.; Yang, J.; Yang, S.; et al. Low CCL19 expression is associated with adverse clinical outcomes for follicular lymphoma patients treated with chemoimmunotherapy. J. Transl. Med. 2021, 19, 399. [Google Scholar] [CrossRef]
- Bradshaw, G.; Haupt, L.M.; Aquino, E.M.; Lea, R.A.; Sutherland, H.G.; Griffiths, L.R. Single Nucleotide Polymorphisms in MIR143 Contribute to Protection against Non-Hodgkin Lymphoma (NHL) in Caucasian Populations. Genes 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.; Wang, X.; Liu, H.; Tian, F.; Song, G. Low expression of miRNA-224 predicts poor clinical outcome in diffuse large B-cell lymphoma treated with R-CHOP. Biomarkers 2015, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Okude, H.; Ori, D.; Kawai, T. Signaling Through Nucleic Acid Sensors and Their Roles in Inflammatory Diseases. Front. Immunol. 2020, 11, 625833. [Google Scholar] [CrossRef] [PubMed]
- Biernat, M.M.; Wróbel, T. Bacterial Infection and Non-Hodgkin B-Cell Lymphoma: Interactions between Pathogen, Host and the Tumor Environment. Int. J. Mol. Sci. 2021, 22, 7372. [Google Scholar] [CrossRef] [PubMed]
- Wotherspoon, A.C.; Ortiz-Hidalgo, C.; Falzon, M.R.; Isaacson, P.G. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991, 338, 1175–1176. [Google Scholar] [CrossRef]
- Wotherspoon, A.C.; Doglioni, C.; Diss, T.C.; Pan, L.; Moschini, A.; de Boni, M.; Isaacson, P.G. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993, 342, 575–577. [Google Scholar] [CrossRef]
- Ferreri, A.J.; Guidoboni, M.; Ponzoni, M.; De Conciliis, C.; Dell’Oro, S.; Fleischhauer, K.; Caggiari, L.; Lettini, A.A.; Dal Cin, E.; Ieri, R.; et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J. Natl. Cancer Inst. 2004, 96, 586–594. [Google Scholar] [CrossRef]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-κB activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [Green Version]
- Vereecke, L.; Beyaert, R.; van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30, 383–391. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Barber, G.N.; Harhaj, E.W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 2010, 285, 14999–15009. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 through gene mutation in B-cell lymphomas. Rinsho Ketsueki 2011, 52, 313–319. [Google Scholar] [PubMed]
- Xagoraris, I.; Farrajota Neves da Silva, P.; Kokaraki, G.; Stathopoulou, K.; Wahlin, B.; Österborg, A.; Herold, N.; Ng, S.B.; Medeiros, L.J.; Drakos, E.; et al. Sting Is Commonly and Differentially Expressed in T- and Nk-Cell but Not B-Cell Non-Hodgkin Lymphomas. Cancers 2022, 14, 1186. [Google Scholar] [CrossRef] [PubMed]
- Rieken, S.; Sassmann, A.; Herroeder, S.; Wallenwein, B.; Moers, A.; Offermanns, S.; Wettschureck, N. G12/G13 family G proteins regulate marginal zone B cell maturation, migration, and polarization. J. Immunol. 2006, 177, 2985–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Liu, Y.; Kunkalla, K.; Singh, R.R.; Blonska, M.; Lin, X.; Agarwal, N.K.; Vega, F. Trimeric G protein-CARMA1 axis links smoothened, the hedgehog receptor transducer, to NF-κB activation in diffuse large B-cell lymphoma. Blood 2013, 121, 4718–4728. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, J.R.; Schmitz, R.; Green, J.A.; Xiao, W.; Larsen, A.B.; Braun, S.E.; An, J.; Xu, Y.; Rosenwald, A.; Ott, G.; et al. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature 2014, 516, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Nogai, H.; Dörken, B.; Lenz, G. Pathogenesis of non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011, 29, 1803–1811. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Sites | |||
|---|---|---|---|
| Subtypes | Nodal (n) | Extranodal (n) | |
| Discovery Cohort | SLL | 20 | 3 |
| FL | 5 | 16 | |
| MCL | 12 | 8 | |
| MZL | 0 | 19 | |
| RL | 15 | 2 | |
| Validation Cohort | SLL | 14 | 6 |
| FL | 53 | 21 | |
| MCL | 13 | 9 | |
| MZL | 5 | 69 | |
| RL | 54 | 38 | |
| Proposed RT-qPCR Assay | IHC and FISH | |
|---|---|---|
| Cost (based on charges in our institution) | USD115-150 (classifier 1) USD230-300 (classifier 1 + 2) | IHC with 7 antibodies: USD605 BCL2 FISH: USD375 B-cell clonality: USD680 |
| Turnaround time | Within a day | IHC: 1–2 days FISH: 5–7 days Clonality analysis: 7–10 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hue, S.S.-S.; Jin, Y.; Cheng, H.; Bin Masroni, M.S.; Tang, L.W.T.; Ho, Y.H.; Ong, D.B.-L.; Leong, S.M.; Tan, S.Y. Tissue-Specific microRNA Expression Profiling to Derive Novel Biomarkers for the Diagnosis and Subtyping of Small B-Cell Lymphomas. Cancers 2023, 15, 453. https://doi.org/10.3390/cancers15020453
Hue SS-S, Jin Y, Cheng H, Bin Masroni MS, Tang LWT, Ho YH, Ong DB-L, Leong SM, Tan SY. Tissue-Specific microRNA Expression Profiling to Derive Novel Biomarkers for the Diagnosis and Subtyping of Small B-Cell Lymphomas. Cancers. 2023; 15(2):453. https://doi.org/10.3390/cancers15020453
Chicago/Turabian StyleHue, Susan Swee-Shan, Yu Jin, He Cheng, Muhammad Sufyan Bin Masroni, Lloyd Wei Tat Tang, Yong Howe Ho, Diana Bee-Lan Ong, Sai Mun Leong, and Soo Yong Tan. 2023. "Tissue-Specific microRNA Expression Profiling to Derive Novel Biomarkers for the Diagnosis and Subtyping of Small B-Cell Lymphomas" Cancers 15, no. 2: 453. https://doi.org/10.3390/cancers15020453
APA StyleHue, S. S. -S., Jin, Y., Cheng, H., Bin Masroni, M. S., Tang, L. W. T., Ho, Y. H., Ong, D. B. -L., Leong, S. M., & Tan, S. Y. (2023). Tissue-Specific microRNA Expression Profiling to Derive Novel Biomarkers for the Diagnosis and Subtyping of Small B-Cell Lymphomas. Cancers, 15(2), 453. https://doi.org/10.3390/cancers15020453