

Mucosal Microbiome in Patients with Early Bowel Polyps: Inferences from Short-Read and Long-Read 16S rRNA Sequencing



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Inclusion and Exclusion Criteria
- Adults ≥ 18 years undergoing colonoscopy for either a positive fecal occult blood test, positive family history of sporadic bowel neoplasia, rectal bleeding, or a scheduled colonic surveillance.
- Written consent to collect tissue for research purposes and to complete a questionnaire on family medical history and general lifestyle choices.
- Adequate tissue for research collection and pathological analysis as specified by the treating pathologist.
- Patients diagnosed with hereditary bowel polyposis diseases including Lynch syndrome (HNPCC) and familiar adenomatous polyposis (FAP).
- Patients with Crohn’s disease, ulcerative colitis, active diverticulitis, or other inflammatory bowel diseases.
- Any condition compromising the patient’s ability to give informed consent or patients who did not consent to the return of incidental findings from molecular analyses.
- Patients who had taken antibiotics up to four weeks before the colonoscopy.
2.3. Specimen Collection
2.4. Bacterial DNA Extraction
2.5. 16S rRNA Gene Sequencing and Bioinformatic Analyses
2.5.1. Short Read Sequencing
2.5.2. PacBio Long-Read Sequencing
2.6. Taxonomy Assignment
2.7. Statistical Analysis
2.7.1. Sequencing Quality
2.7.2. Alpha Diversity Analyses
2.7.3. Beta Diversity Analyses
2.8. PERMANOVA
2.9. Differential Abundance Analyses
2.10. Multivariate Analyses
3. Results
3.1. Participant Clinical Data
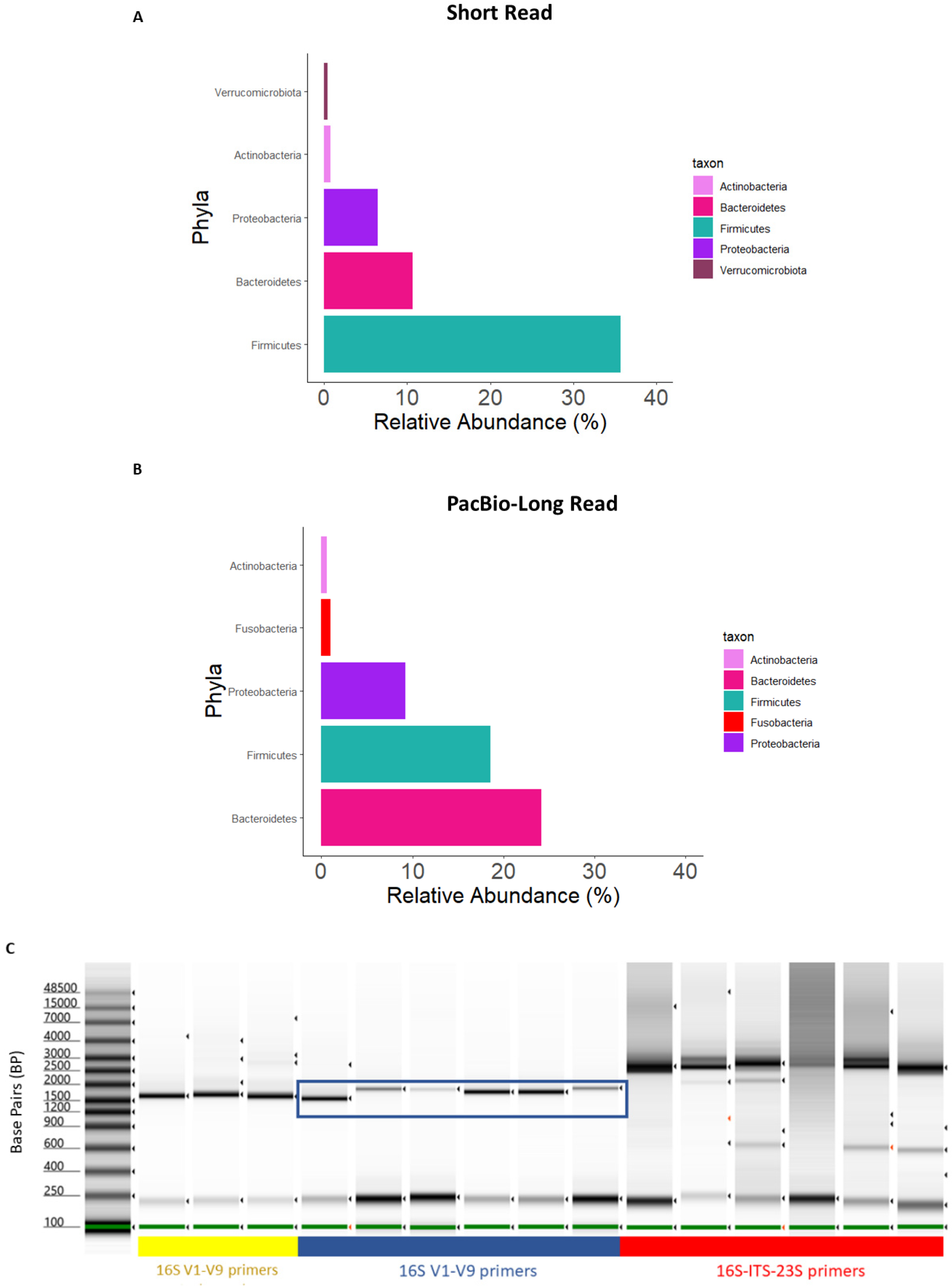
3.2. Comparison of Long-Read Primer Sets
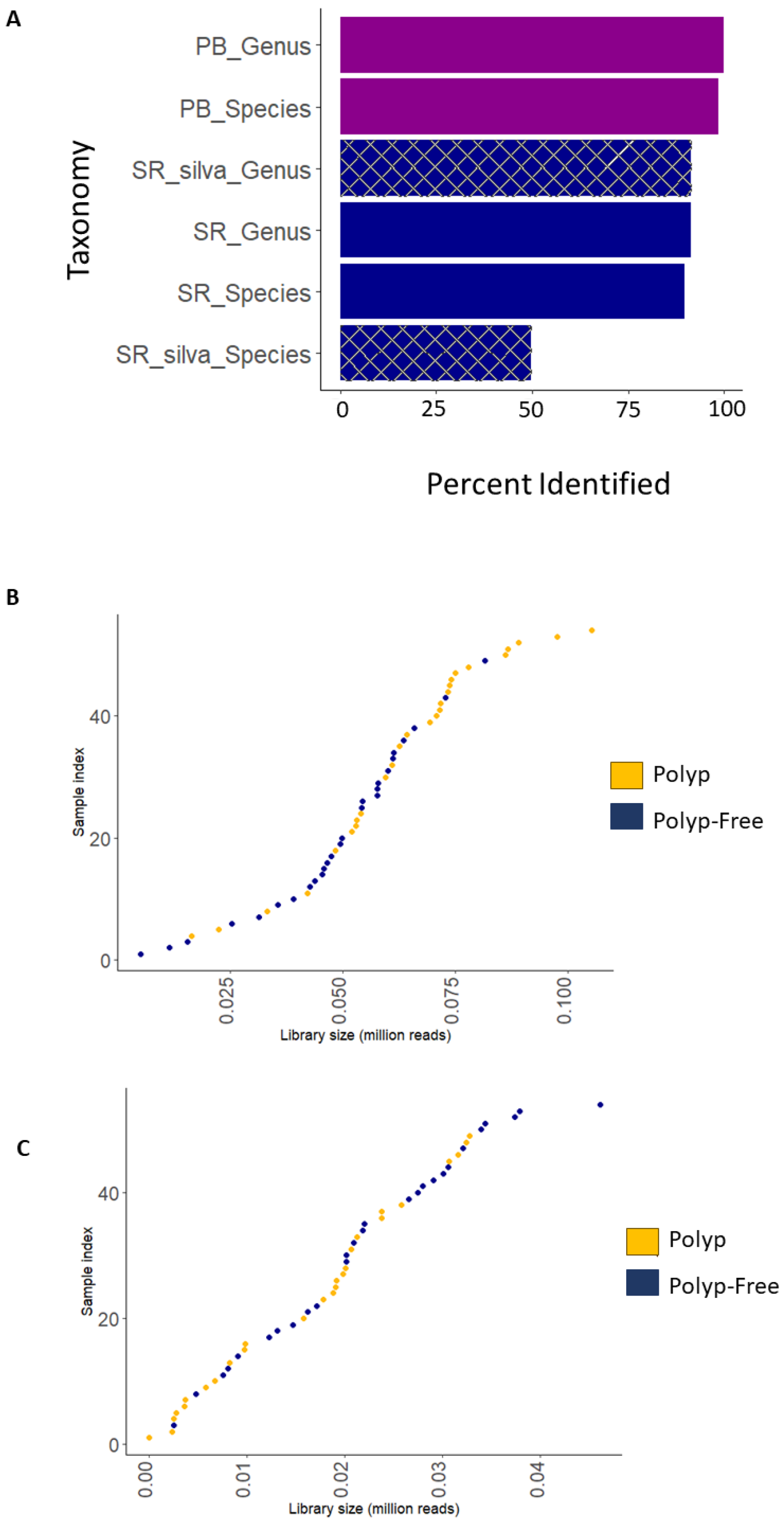
3.3. Comparison of Short-Read and Long-Read Sequencing Quality
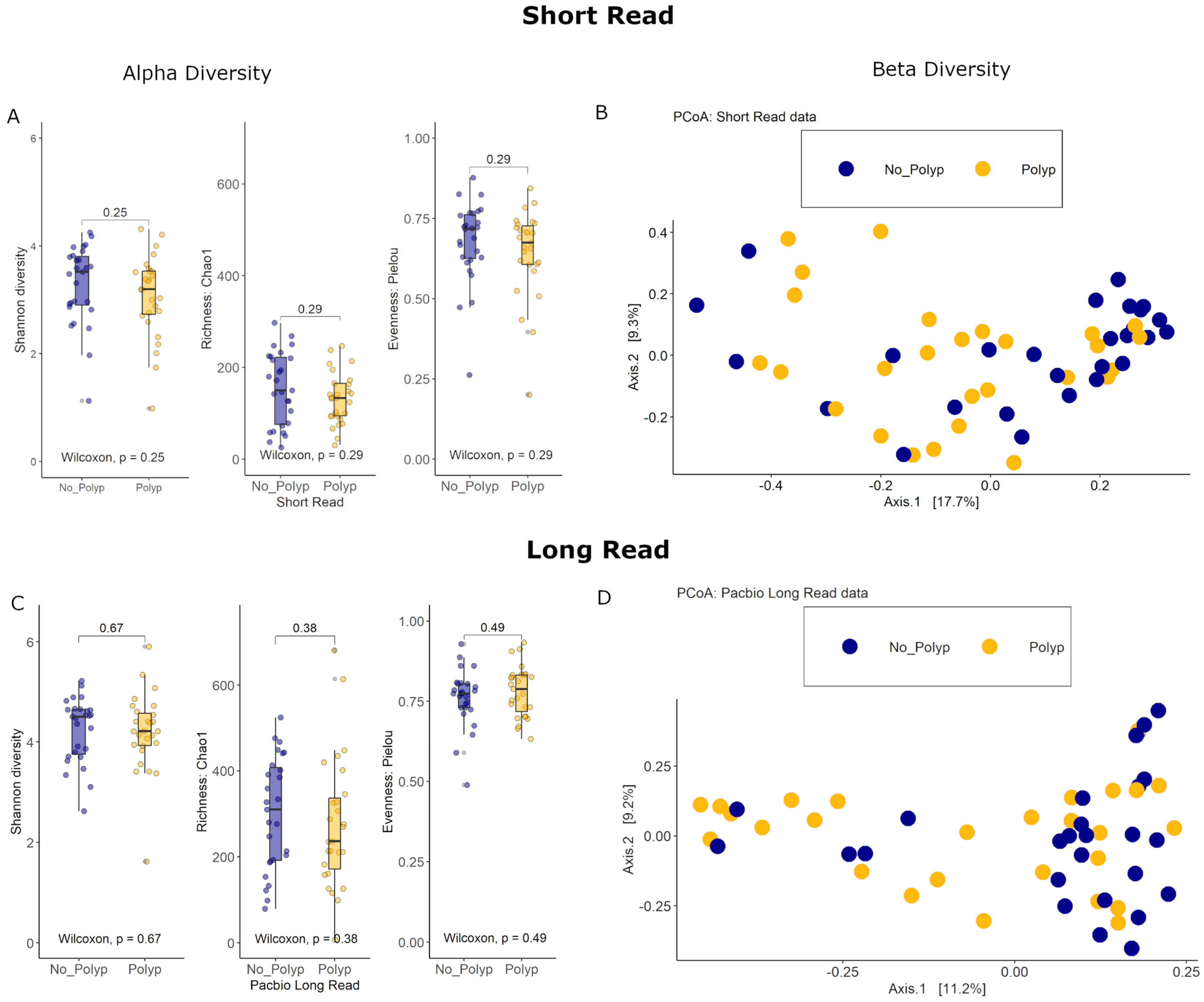
3.4. Alpha Diversity Analyses
3.5. Beta Diversity Analyses
3.6. PERMANOVA
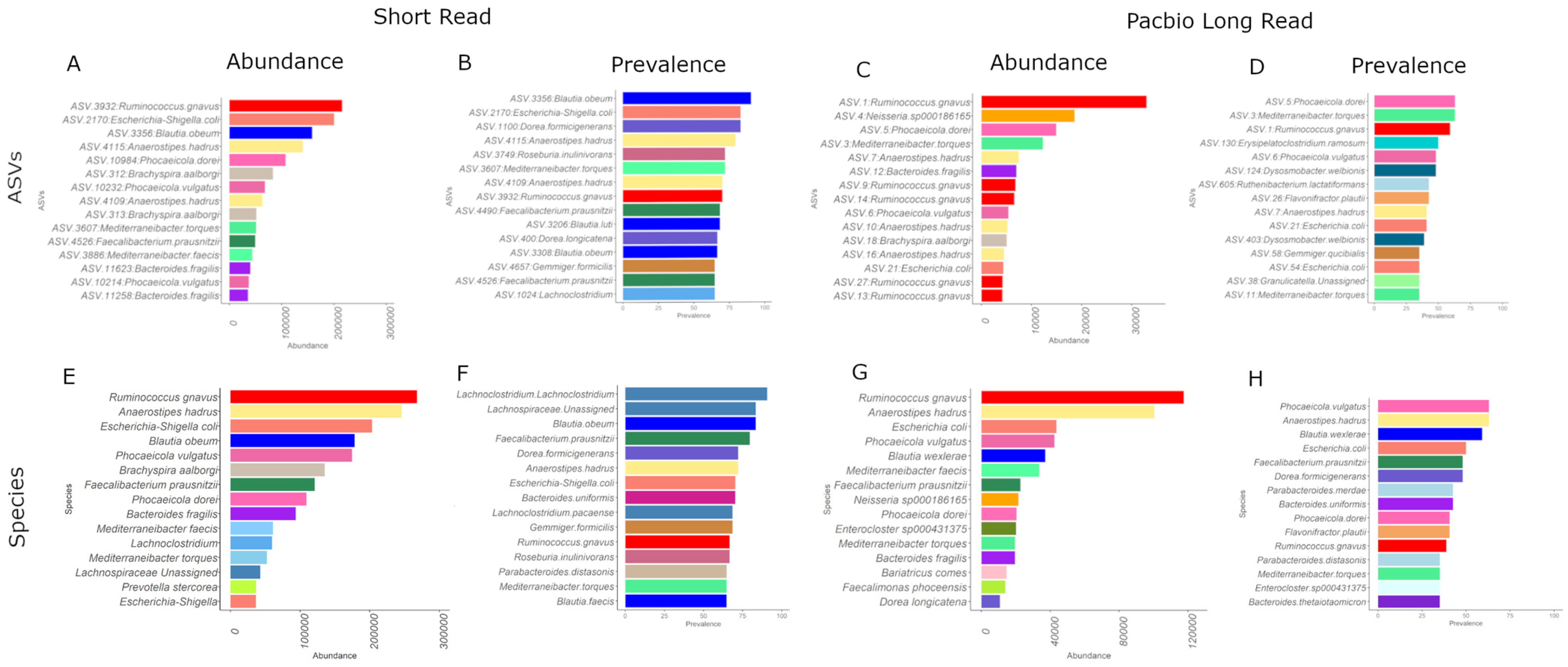
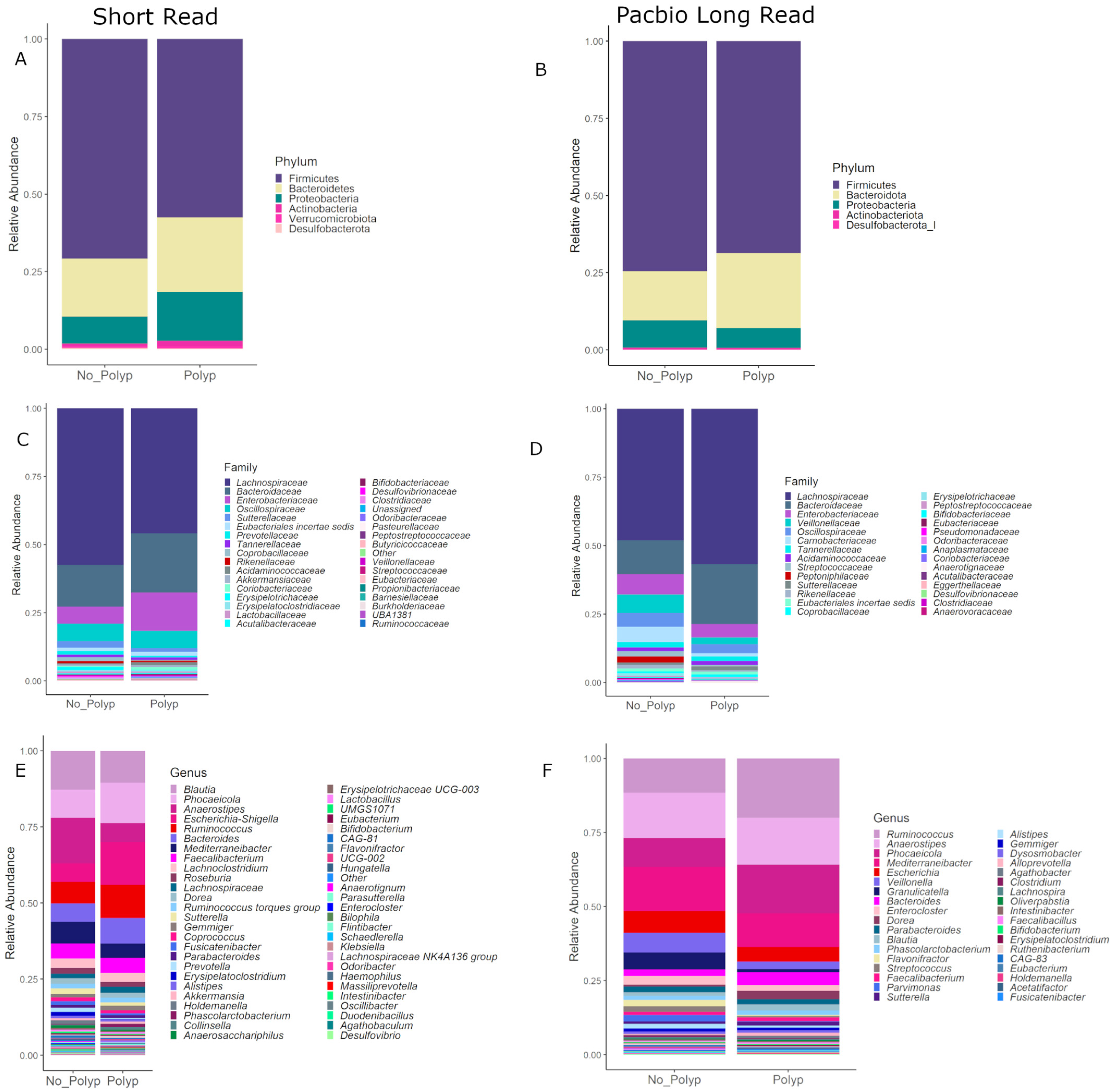
3.7. Community Composition
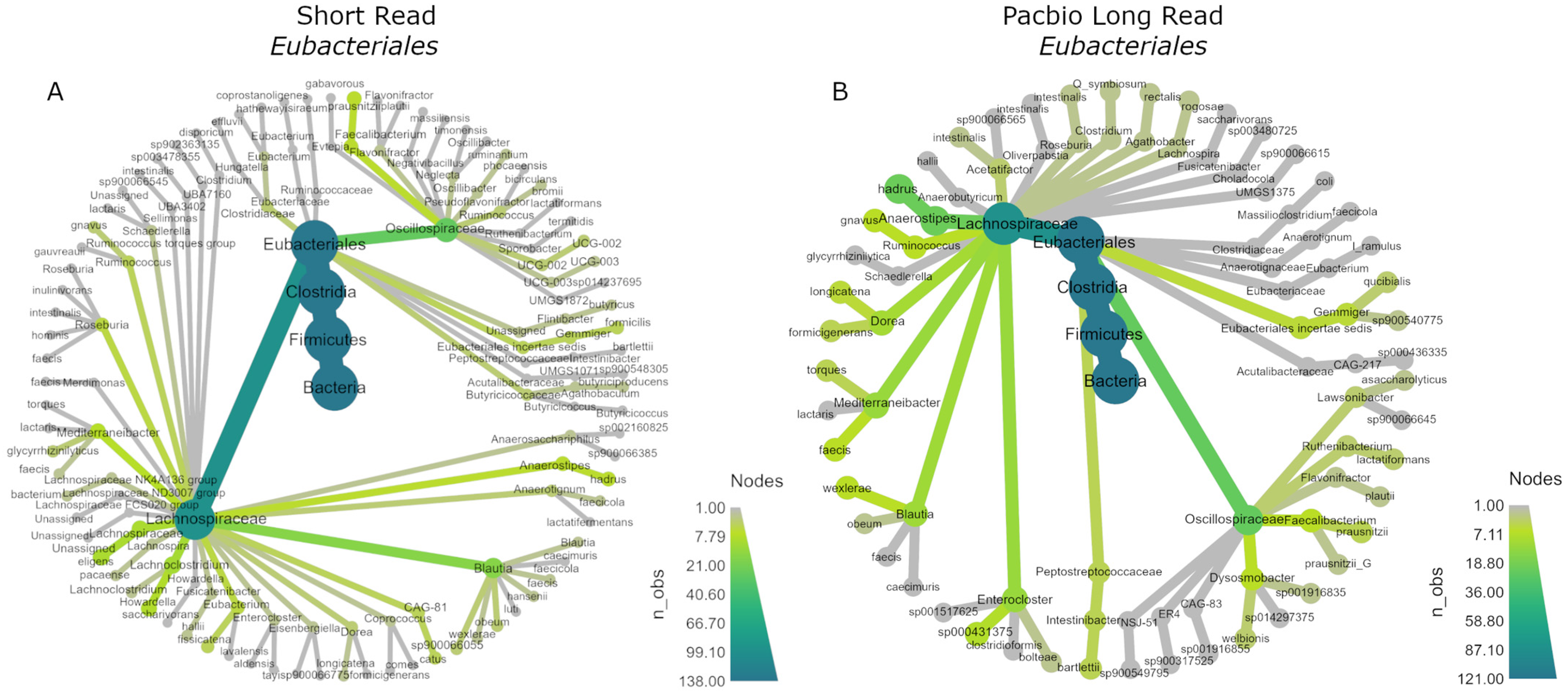
3.8. Phylogenetic Trees
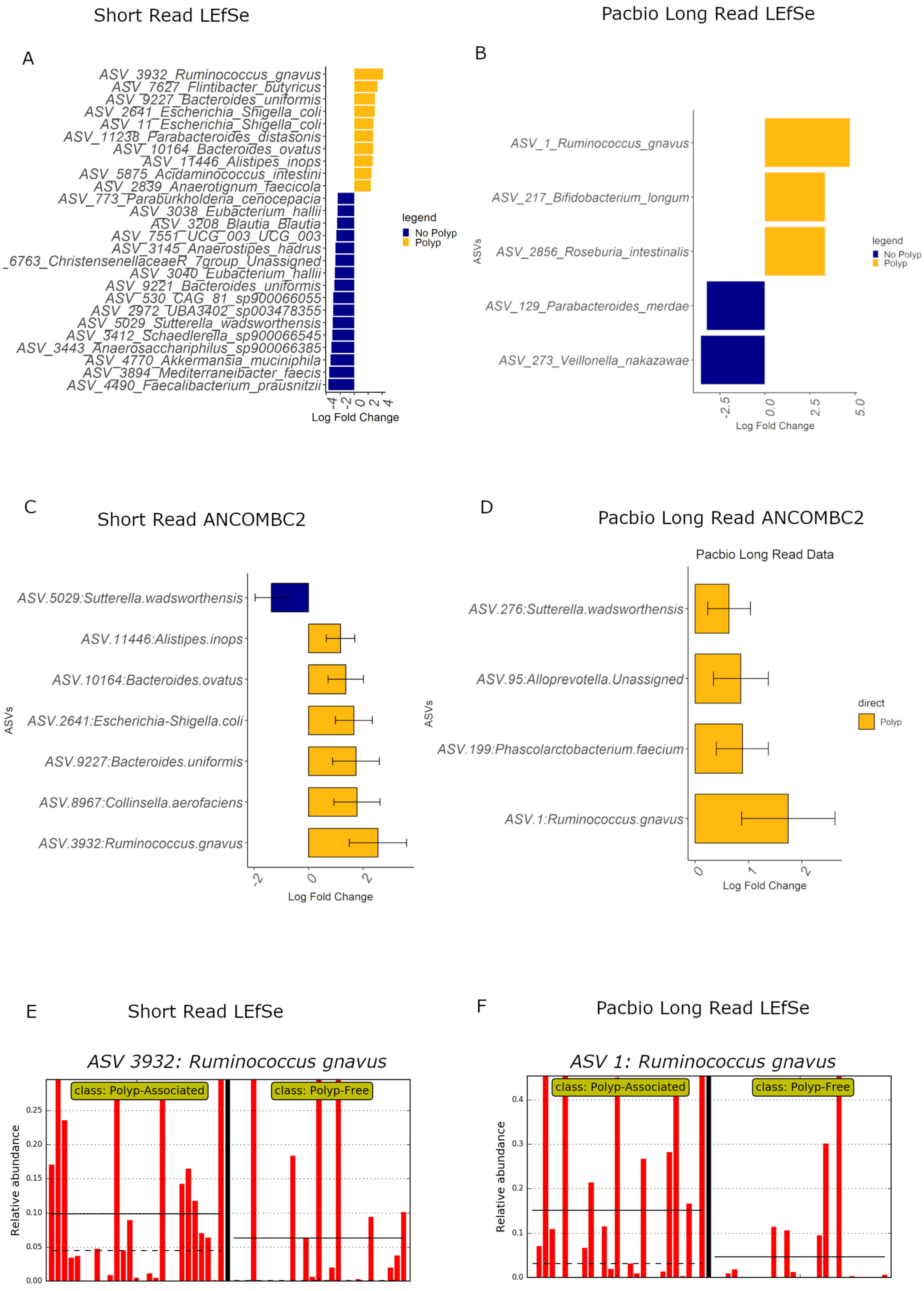
3.9. Differential Abundance Analyses
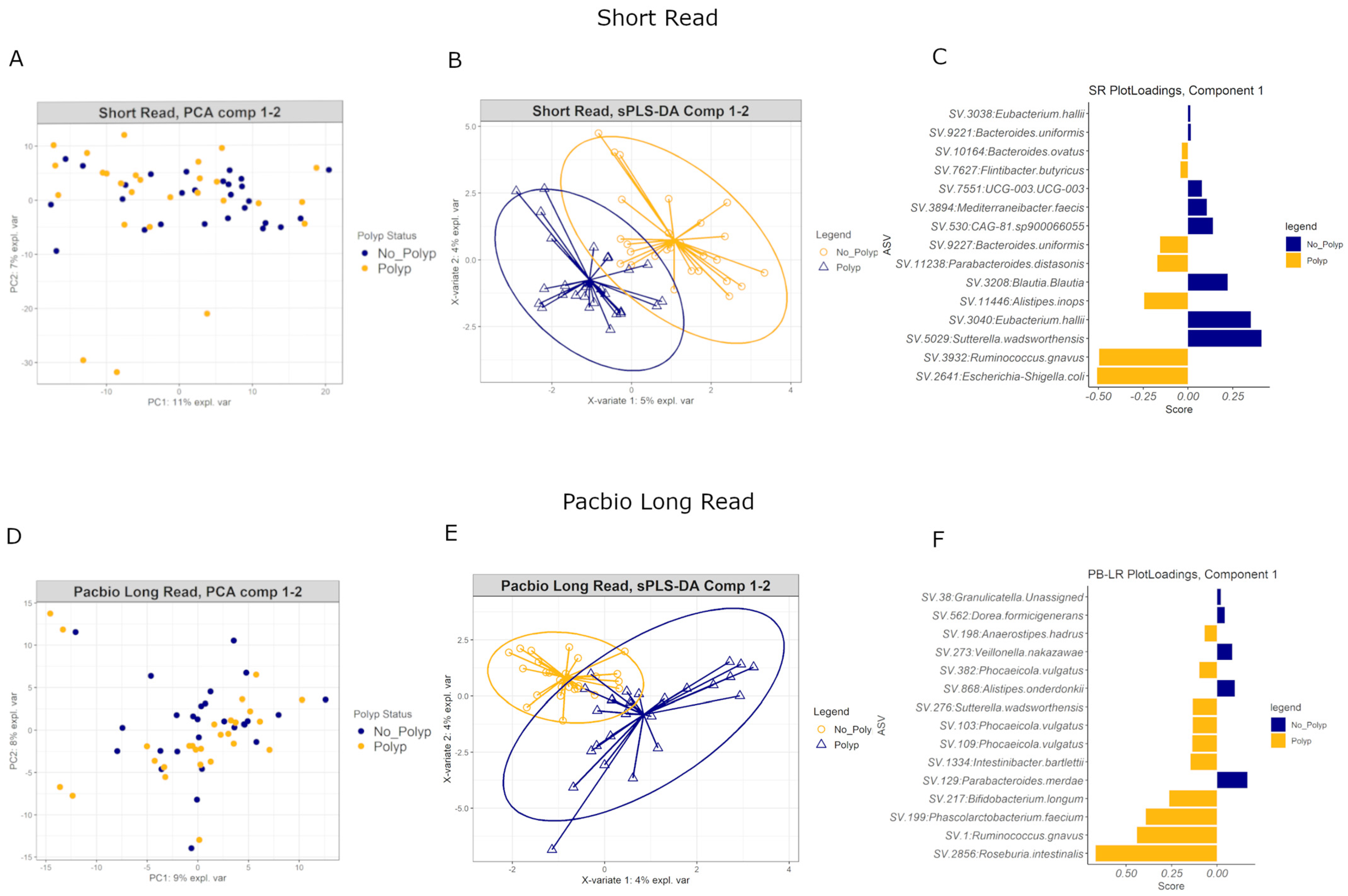
3.10. Multivariate Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef]
- Abed, J.; Emgard, J.E.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Zhou, R.; Ng, S.C.; Li, J.; Huang, M.; Zhou, F.; Wang, X.; Shen, B.; Kamm, M.A.; et al. Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine 2014, 93, e51. [Google Scholar] [CrossRef]
- Shen, X.J.; Rawls, J.F.; Randall, T.; Burcal, L.; Mpande, C.N.; Jenkins, N.; Jovov, B.; Abdo, Z.; Sandler, R.S.; Keku, T.O. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 2010, 1, 138–147. [Google Scholar] [CrossRef]
- Durban, A.; Abellan, J.J.; Jimenez-Hernandez, N.; Ponce, M.; Ponce, J.; Sala, T.; D’Auria, G.; Latorre, A.; Moya, A. Assessing gut microbial diversity from feces and rectal mucosa. Microb. Ecol. 2011, 61, 123–133. [Google Scholar] [CrossRef]
- Ringel, Y.; Maharshak, N.; Ringel-Kulka, T.; Wolber, E.A.; Sartor, R.B.; Carroll, I.M. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes 2015, 6, 173–181. [Google Scholar] [CrossRef]
- Mira-Pascual, L.; Cabrera-Rubio, R.; Ocon, S.; Costales, P.; Parra, A.; Suarez, A.; Moris, F.; Rodrigo, L.; Mira, A.; Collado, M.C. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2015, 50, 167–179. [Google Scholar] [CrossRef]
- Xu, K.; Jiang, B. Analysis of Mucosa-Associated Microbiota in Colorectal Cancer. Med. Sci. Monit. 2017, 23, 4422–4430. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar] [CrossRef]
- Sanapareddy, N.; Legge, R.M.; Jovov, B.; McCoy, A.; Burcal, L.; Araujo-Perez, F.; Randall, T.A.; Galanko, J.; Benson, A.; Sandler, R.S.; et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012, 6, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Song, Q.; Tang, X.; Liang, X.; Fan, H.; Peng, H.; Guo, Q.; Zhang, Z. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Zhou, Y.L.; He, J.; Feng, Z.Q.; Zhang, L.; Lai, X.B.; Zhou, J.X.; Wang, H. Characterizing the composition of intestinal microflora by 16S rRNA gene sequencing. World J. Gastroenterol. 2020, 26, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H.; et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef]
- Dadkhah, E.; Sikaroodi, M.; Korman, L.; Hardi, R.; Baybick, J.; Hanzel, D.; Kuehn, G.; Kuehn, T.; Gillevet, P.M. Gut microbiome identifies risk for colorectal polyps. BMJ Open Gastroenterol. 2019, 6, e000297. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Sears, C.L. Enterotoxigenic Bacteroides fragilis: A rogue among symbiotes. Clin. Microbiol. Rev. 2009, 22, 349–369. [Google Scholar] [CrossRef]
- Wei, P.L.; Hung, C.S.; Kao, Y.W.; Lin, Y.C.; Lee, C.Y.; Chang, T.H.; Shia, B.C.; Lin, J.C. Classification of Changes in the Fecal Microbiota Associated with Colonic Adenomatous Polyps Using a Long-Read Sequencing Platform. Genes 2020, 11, 1374. [Google Scholar] [CrossRef]
- Taylor, W.S.; Pearson, J.; Miller, A.; Schmeier, S.; Frizelle, F.A.; Purcell, R.V. MinION Sequencing of colorectal cancer tumour microbiomes-A comparison with amplicon-based and RNA-Sequencing. PLoS ONE 2020, 15, e0233170. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Application for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2012. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- D’Argenio, V.; Salvatore, F. The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 2015, 451 Pt A, 97–102. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Rogers, M.A.; Ruffin, M.T.t.; Schloss, P.D. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. 2014, 7, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed]
- Rezasoltani, S.; Asadzadeh Aghdaei, H.; Dabiri, H.; Akhavan Sepahi, A.; Modarressi, M.H.; Nazemalhosseini Mojarad, E. The association between fecal microbiota and different types of colorectal polyp as precursors of colorectal cancer. Microb. Pathog. 2018, 124, 244–249. [Google Scholar] [CrossRef]
- Hong, B.Y.; Ideta, T.; Lemos, B.S.; Igarashi, Y.; Tan, Y.; DiSiena, M.; Mo, A.; Birk, J.W.; Forouhar, F.; Devers, T.J.; et al. Characterization of Mucosal Dysbiosis of Early Colonic Neoplasia. NPJ Precis. Oncol. 2019, 3, 29. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef]
- Su, Q.; Liu, Q.; Lau, R.I.; Zhang, J.; Xu, Z.; Yeoh, Y.K.; Leung, T.W.H.; Tang, W.; Zhang, L.; Liang, J.Q.Y.; et al. Faecal microbiome-based machine learning for multi-class disease diagnosis. Nat. Commun. 2022, 13, 6818. [Google Scholar] [CrossRef]
- Moschen, A.R.; Gerner, R.R.; Wang, J.; Klepsch, V.; Adolph, T.E.; Reider, S.J.; Hackl, H.; Pfister, A.; Schilling, J.; Moser, P.L.; et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host Microbe 2016, 19, 455–469. [Google Scholar] [CrossRef]
- Hajjar, R.; Richard, C.S.; Santos, M.M. The role of butyrate in surgical and oncological outcomes in colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G601–G608. [Google Scholar] [CrossRef]
- Xi, Y.; Jing, Z.; Wei, W.; Chun, Z.; Quan, Q.; Qing, Z.; Jiamin, X.; Shuwen, H. Inhibitory effect of sodium butyrate on colorectal cancer cells and construction of the related molecular network. BMC Cancer 2021, 21, 127. [Google Scholar] [CrossRef]
- Coradini, D.; Pellizzaro, C.; Marimpietri, D.; Abolafio, G.; Daidone, M.G. Sodium butyrate modulates cell cycle-related proteins in HT29 human colonic adenocarcinoma cells. Cell Prolif. 2000, 33, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Li, Z.; Mao, L.; Chen, S.; Sun, S. Sodium butyrate induces autophagy in colorectal cancer cells through LKB1/AMPK signaling. J. Physiol. Biochem. 2019, 75, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Barone, M.; Mosca, A.; Viggiani, M.T.; Di Leo, A.; Debellis, L.; Troisi, M.; Daniele, A.; Santacroce, L. Gut microbiota imbalance is related to sporadic colorectal neoplasms. A pilot study. Appl. Sci. 2019, 9, 5491. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jiao, N.; Zhu, R.; Zhang, Y.; Wu, D.; Wang, A.J.; Fang, S.; Tao, L.; Li, Y.; Cheng, S.; et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat. Commun. 2021, 12, 3063. [Google Scholar] [CrossRef] [PubMed]
- Mashima, I.; Theodorea, C.F.; Djais, A.A.; Kunihiro, T.; Kawamura, Y.; Otomo, M.; Saitoh, M.; Tamai, R.; Kiyoura, Y. Veillonella nakazawae sp. nov., an anaerobic Gram-negative coccus isolated from the oral cavity of Japanese children. Int. J. Syst. Evol. Microbiol. 2021, 71, 004583. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chuang, C.H.; Miao, Z.F.; Yip, K.L.; Liu, C.J.; Li, L.H.; Wu, D.C.; Cheng, T.L.; Lin, C.Y.; Wang, J.Y. Gut microbiota composition in chemotherapy and targeted therapy of patients with metastatic colorectal cancer. Front. Oncol. 2022, 12, 955313. [Google Scholar] [CrossRef]
- Hale, V.L.; Chen, J.; Johnson, S.; Harrington, S.C.; Yab, T.C.; Smyrk, T.C.; Nelson, H.; Boardman, L.A.; Druliner, B.R.; Levin, T.R.; et al. Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer Epidemiol. Biomark. Prev. 2017, 26, 85–94. [Google Scholar] [CrossRef]
- Singh, J.; Rivenson, A.; Tomita, M.; Shimamura, S.; Ishibashi, N.; Reddy, B.S. Bifidobacterium longum, a lactic acid-producing intestinal bacterium inhibits colon cancer and modulates the intermediate biomarkers of colon carcinogenesis. Carcinogenesis 1997, 18, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, C.A.; Gamal-Eldeen, A.M.; El-Hussieny, E.A.; Raafat, B.M.; Mehanna, N.S.; Talaat, R.M.; Shaaban, M.T. Bifidobacterium longum Suppresses Murine Colorectal Cancer through the Modulation of oncomiRs and Tumor Suppressor miRNAs. Nutr. Cancer 2019, 71, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Shrode, R.L.; Knobbe, J.E.; Cady, N.; Yadav, M.; Hoang, J.; Cherwin, C.; Curry, M.; Garje, R.; Vikas, P.; Sugg, S. Breast cancer patients from the Midwest region of the United States have reduced levels of short-chain fatty acid-producing gut bacteria. Sci. Rep. 2023, 13, 526. [Google Scholar] [CrossRef]
- Hasan, R.; Bose, S.; Roy, R.; Paul, D.; Rawat, S.; Nilwe, P.; Chauhan, N.K.; Choudhury, S. Tumor tissue-specific bacterial biomarker panel for colorectal cancer: Bacteroides massiliensis, Alistipes species, Alistipes onderdonkii, Bifidobacterium pseudocatenulatum, Corynebacterium appendicis. Arch. Microbiol. 2022, 204, 348. [Google Scholar] [CrossRef]
- Masoodi, I.; Alshanqeeti, A.S.; Alyamani, E.J.; AlLehibi, A.A.; Alqutub, A.N.; Alsayari, K.N.; Alomair, A.O. Microbial dysbiosis in irritable bowel syndrome: A single-center metagenomic study in Saudi Arabia. JGH Open 2020, 4, 649–655. [Google Scholar] [CrossRef]
- Khannous-Lleiffe, O.; Willis, J.R.; Saus, E.; Moreno, V.; Castellvi-Bel, S.; Gabaldon, T.; on behalf of the CRIPREV Consortium. Microbiome Profiling from Fecal Immunochemical Test Reveals Microbial Signatures with Potential for Colorectal Cancer Screening. Cancers 2022, 15, 120. [Google Scholar] [CrossRef]
- Ren, X.; Xu, J.; Zhang, Y.; Chen, G.; Zhang, Y.; Huang, Q.; Liu, Y. Bacterial Alterations in Post-Cholecystectomy Patients Are Associated With Colorectal Cancer. Front. Oncol. 2020, 10, 1418. [Google Scholar] [CrossRef]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; Andreu-Sanchez, S.; Chen, L.; Collij, V.; Hu, S.; et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 2022, 604, 732–739. [Google Scholar] [CrossRef]
- Wei, P.L.; Hung, C.S.; Kao, Y.W.; Lin, Y.C.; Lee, C.Y.; Chang, T.H.; Shia, B.C.; Lin, J.C. Characterization of Fecal Microbiota with Clinical Specimen Using Long-Read and Short-Read Sequencing Platform. Int. J. Mol. Sci. 2020, 21, 7110. [Google Scholar] [CrossRef]
- Bang, E.; Oh, S.; Ju, U.; Chang, H.E.; Hong, J.S.; Baek, H.J.; Kim, K.S.; Lee, H.J.; Park, K.U. Factors influencing oral microbiome analysis: From saliva sampling methods to next-generation sequencing platforms. Sci. Rep. 2023, 13, 10086. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Polyp Patients | Polyp-Free Patients | p-Value | ||
|---|---|---|---|---|---|
| N | 27 | 27 | |||
| Age | 68 (27–86) 1 | 57 (37,82) 1 | 0.11 2 | ||
| Gender | Female | 9 | 9 | 0.8 3 | |
| Male | 18 | 18 | |||
| BMI | 25 (19,30) 1 | 25 (23,31) 1 | 0.2 2 | ||
| Type of Lesion | SL | <10mm | 1 | 0 | |
| ≥10mm | 0 | 0 | |||
| TA | <10mm | 22 | 0 | ||
| ≥10mm | 2 | 0 | |||
| TVA | <10mm | 0 | 0 | ||
| ≥10mm | 2 | 0 | |||
| Indications for colonoscopy | Surveillance—1st degree relative Polyp or CRC | 2 | 4 | 0.7 3 | |
| Surveillance—Personal History Polyp or CRC | 11 | 8 | |||
| Rectal bleeding | 7 | 10 | |||
| FOBT+/FIT+ | 4 | 2 | |||
| Abnormal Bowel Symptoms | 2 | 1 | |||
| Other | 1 | 2 |
| Sequencing Counts | Short Read | PacBio Long Read |
|---|---|---|
| Total | 20,352,406 | 4,784,897 |
| Average (median) per sample | 96,504 (97,959) | 27,367 (27,545) |
| Average (median) per sample—post-filter, non-chimeric | 61,305 (60,287) | 19,205 (19,999) |
| Average (median) per sample—post filter, non-chimeric (%) | 64.16 (63.29) | 72.25 (78.42) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welham, Z.; Li, J.; Engel, A.F.; Molloy, M.P. Mucosal Microbiome in Patients with Early Bowel Polyps: Inferences from Short-Read and Long-Read 16S rRNA Sequencing. Cancers 2023, 15, 5045. https://doi.org/10.3390/cancers15205045
Welham Z, Li J, Engel AF, Molloy MP. Mucosal Microbiome in Patients with Early Bowel Polyps: Inferences from Short-Read and Long-Read 16S rRNA Sequencing. Cancers. 2023; 15(20):5045. https://doi.org/10.3390/cancers15205045
Chicago/Turabian StyleWelham, Zoe, Jun Li, Alexander F. Engel, and Mark P. Molloy. 2023. "Mucosal Microbiome in Patients with Early Bowel Polyps: Inferences from Short-Read and Long-Read 16S rRNA Sequencing" Cancers 15, no. 20: 5045. https://doi.org/10.3390/cancers15205045
APA StyleWelham, Z., Li, J., Engel, A. F., & Molloy, M. P. (2023). Mucosal Microbiome in Patients with Early Bowel Polyps: Inferences from Short-Read and Long-Read 16S rRNA Sequencing. Cancers, 15(20), 5045. https://doi.org/10.3390/cancers15205045