
Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Cytogenetics Data
2.3. Next-Generation Sequencing
2.4. Data and Statistical Analysis
3. Results
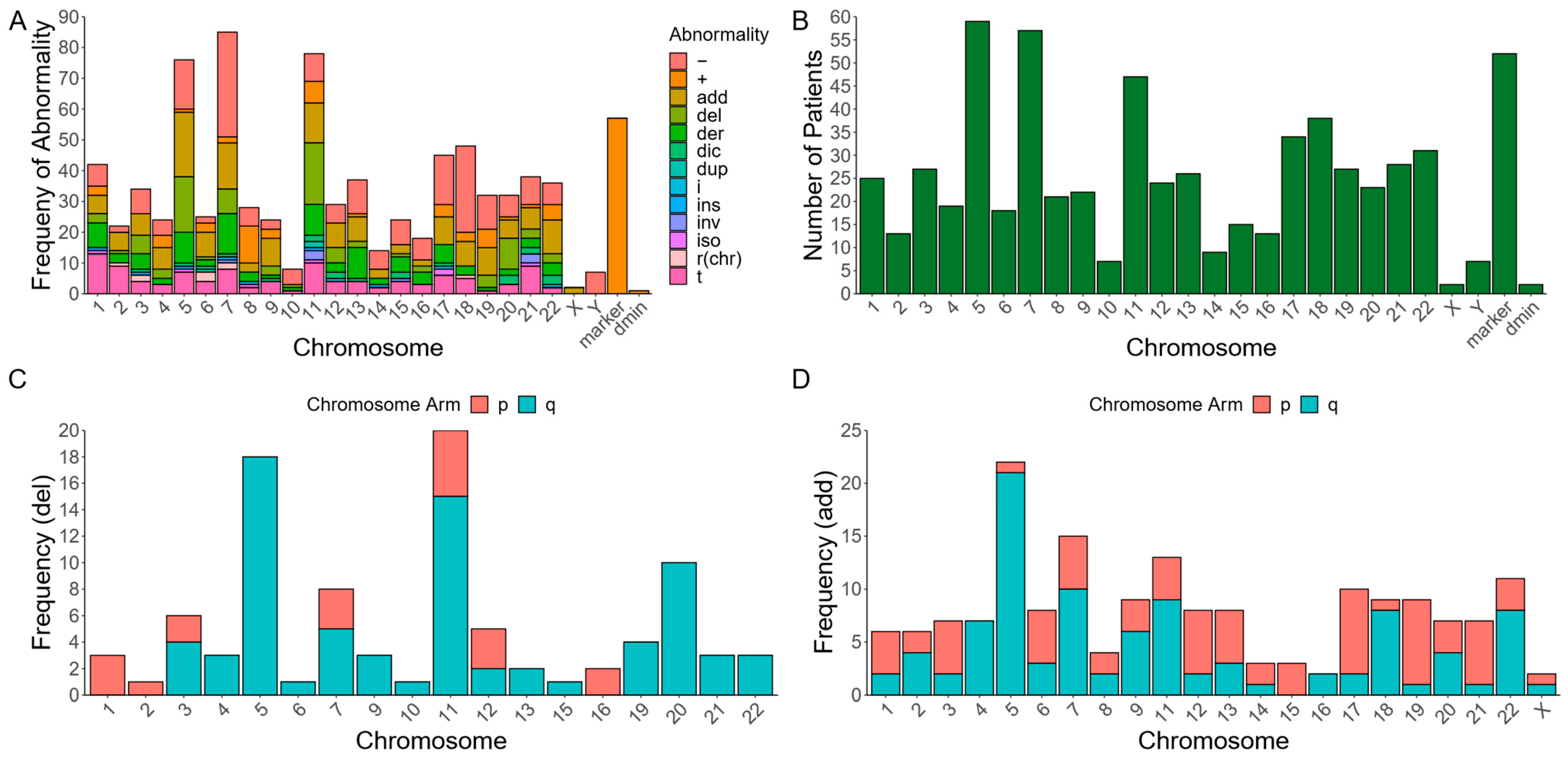
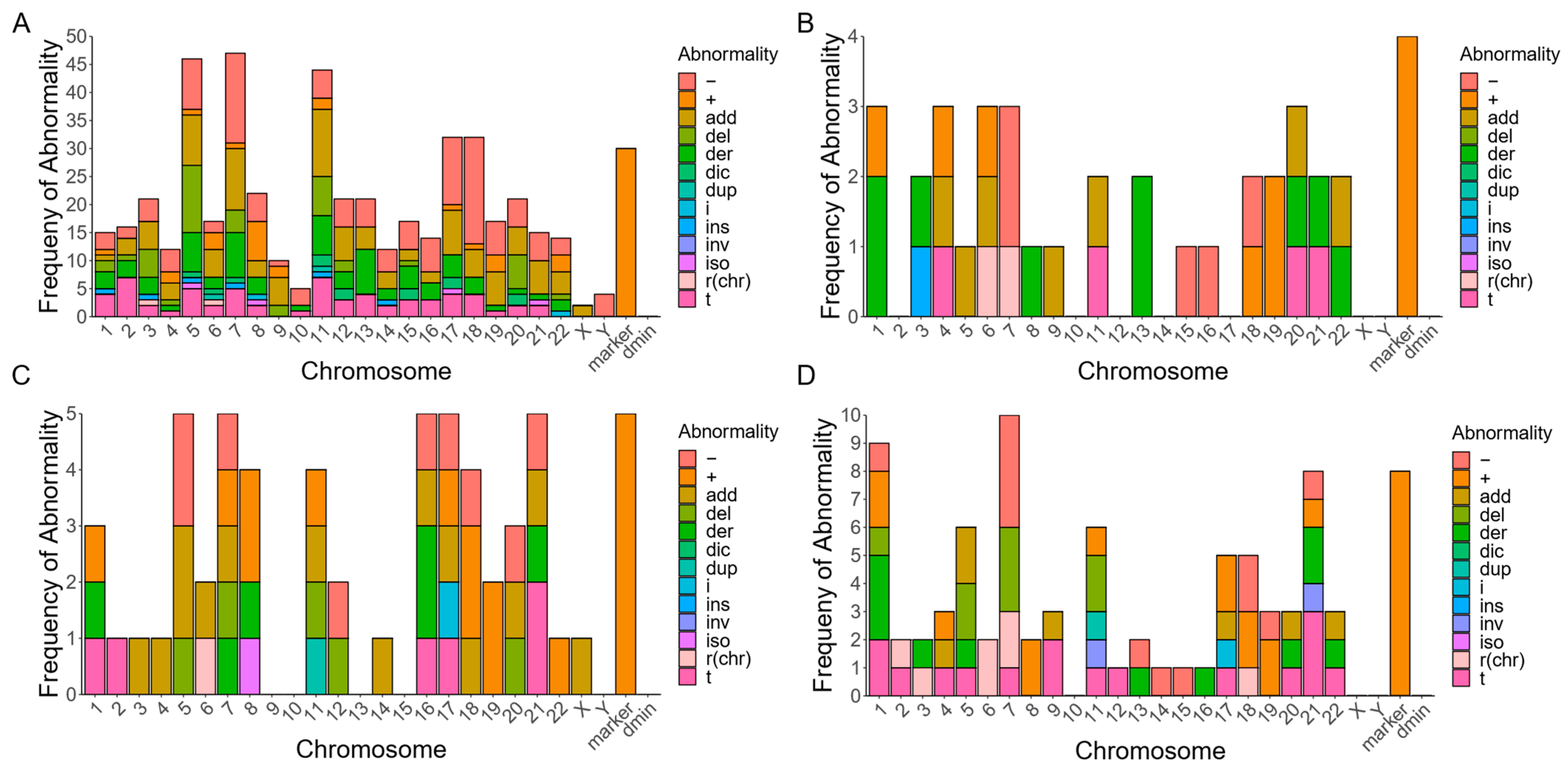
3.1. Subtypes of RCs in Myeloid Malignancies with Various Chromosomal Abnormalities
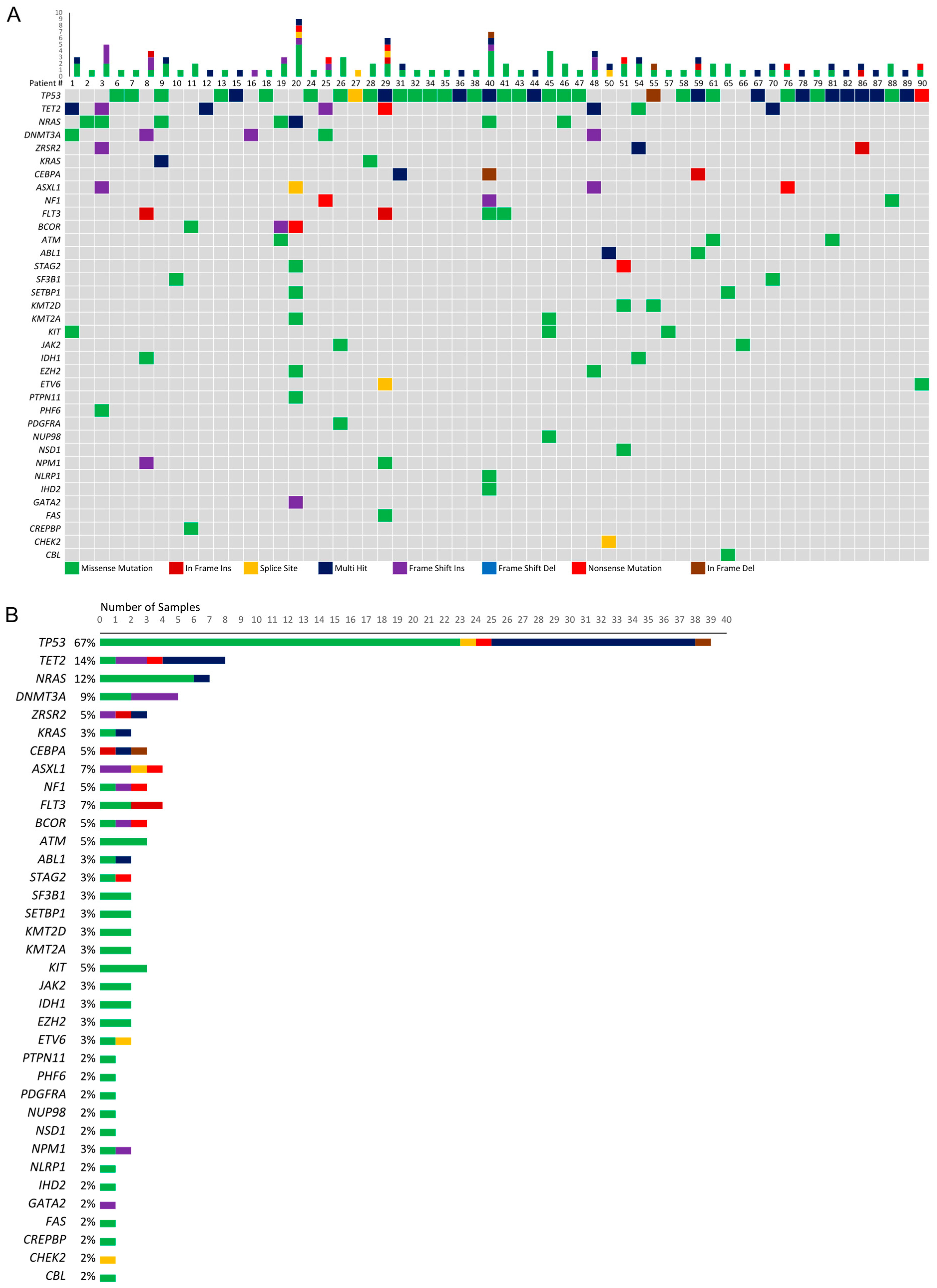
3.2. Genetic Mutations among Patients with Myeloid Malignancies and RCs
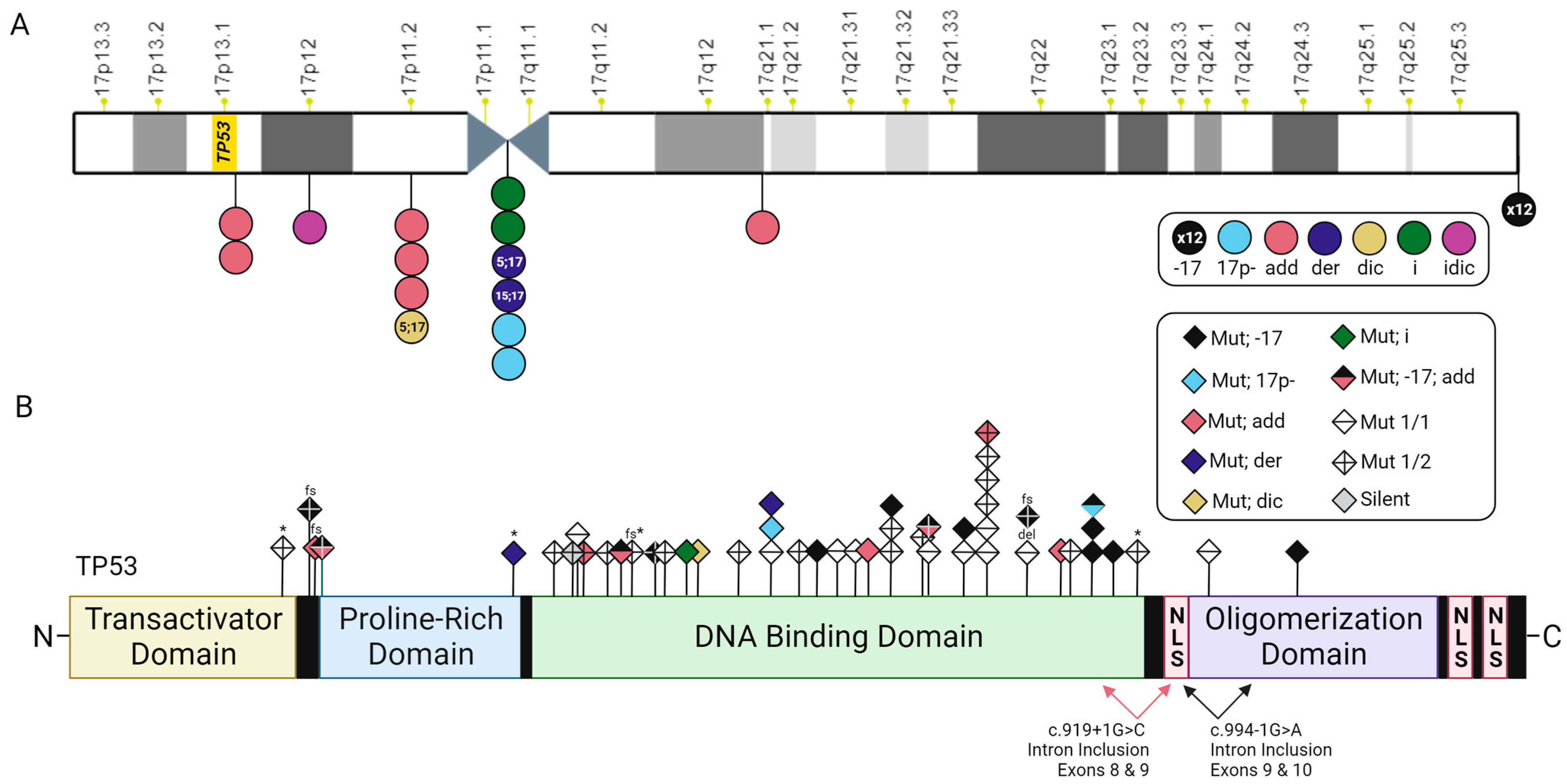
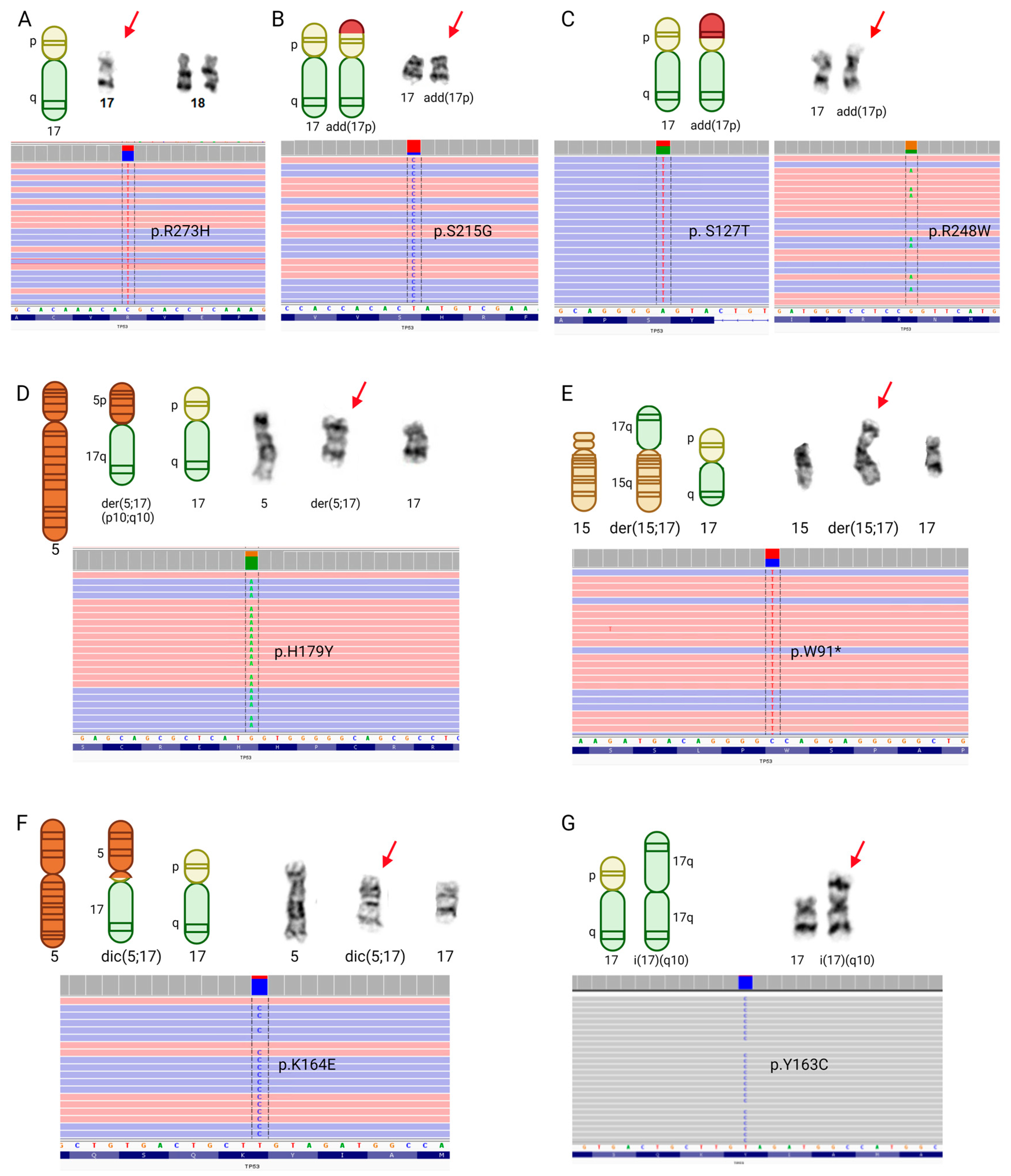
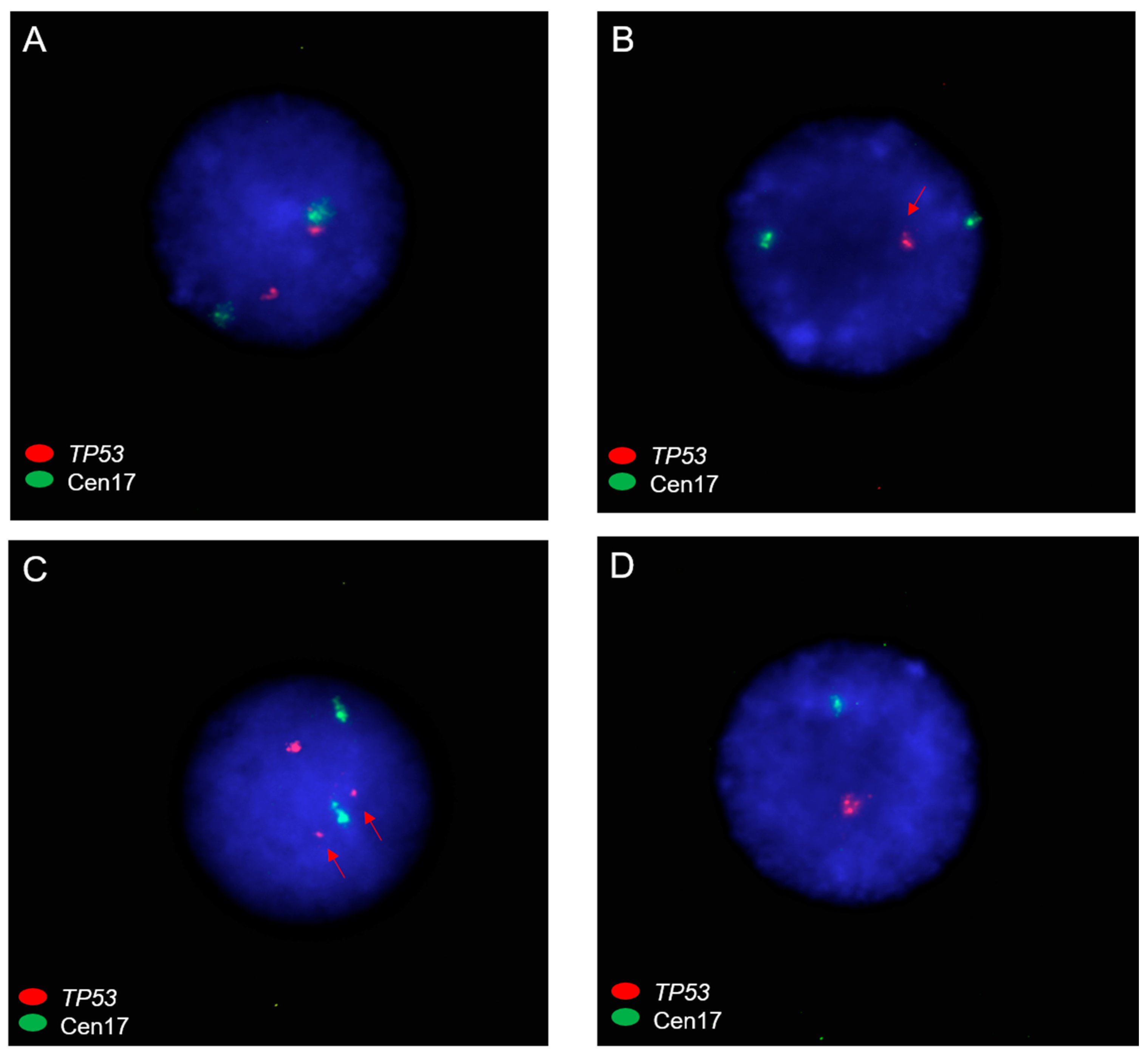
3.3. TP53 Mutations and Chromosome 17 Abnormalities among Patients with Myeloid Malignancies and RCs
3.4. Subtypes of RCs in Lymphoid Malignancies with Various Chromosomal Abnormalities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yip, M.-Y. Autosomal Ring Chromosomes in Human Genetic Disorders. Transl. Pediatr. 2015, 4, 164. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, R.S.; Meloni, V.F.A.; Kim, C.A.; Pellegrino, R.; Takeno, S.S.; Spinner, N.B.; Conlin, L.K.; Christofolini, D.M.; Kulikowski, L.D.; Melaragno, M.I. Mechanisms of Ring Chromosome Formation, Ring Instability and Clinical Consequences. BMC Med. Genet. 2011, 12, 171. [Google Scholar] [CrossRef] [PubMed]
- Kjessler, B.; Gustavson, K.-H.; Wigertz, A. Apparently Non-Deleted Ring-1 Chromosome and Extreme Growth Failure in a Mentally Retarded Girl. Clin. Genet. 1978, 14, 8–15. [Google Scholar] [CrossRef]
- McGinniss, M.J.; Kazazian, H.H.; Stetten, G.; Petersen, M.B.; Boman, H.; Engel, E.; Greenberg, F.; Hertz, J.M.; Johnson, A.; Laca, Z.; et al. Mechanisms of Ring Chromosome Formation in 11 Cases of Human Ring Chromosome 21. Am. J. Hum. Genet. 1992, 50, 15–28. [Google Scholar]
- Mcginniss, M.J.; Brown, D.H.; Burke, L.W.; Mascarello, J.T.; Jones, M.C. Ring Chromosome X in a Child With Manifestations of Kabuki Syndrome. J. Med. Genet. 1997, 70, 37–42. [Google Scholar] [CrossRef]
- Li, P.; Dupont, B.; Hu, Q.; Crimi, M.; Shen, Y.; Lebedev, I.; Liehr, T. The Past, Present, and Future for Constitutional Ring Chromosomes: A Report of the International Consortium for Human Ring Chromosomes. Hum. Genet. Genom. Adv. 2022, 3, 100139. [Google Scholar] [CrossRef]
- Pristyazhnyuk, I.E.; Menzorov, A.G. Ring Chromosomes: From Formation to Clinical Potential. Protoplasma 2018, 255, 439–449. [Google Scholar] [CrossRef]
- Gebhart, E. Ring Chromosomes in Human Neoplasias. Cytogenet. Genome Res. 2008, 121, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Höglund, M.; Mertens, F.; Johansson, B.; Dal Cin, P.; Van Den Berghe, H.; Earnshaw, W.C.; Mitelman, F.; Mandahl, N. The Structure and Dynamics of Ring Chromosomes in Human Neoplastic and Non-Neoplastic Cells. Hum. Genet. 1999, 104, 315–325. [Google Scholar] [CrossRef]
- Park, T.S.; Kim, J.; Song, J.; Song, S.; Suh, B.; Choi, J.R.; Kim, S.J.; Lee, H.W.; Min, Y.H. Association between Acute Promyelocytic Leukemia and Ring Chromosome 6. Cancer Genet. Cytogenet. 2009, 192, 48–50. [Google Scholar] [CrossRef]
- Werner-Favre, C.; Beris, P.; Piguet, D.; Engel, E. Ring Chromosomes and Hematologic Disorders. Cancer Genet. Cytogenet. 1986, 23, 265–267. [Google Scholar] [CrossRef]
- Chandran, R.K.; Geetha, N.; Sakthivel, K.M.; Kumar, R.S.; Krishna, K.M.N.J.; Sreedharan, H. Impact of Additional Chromosomal Aberrations on the Disease Progression of Chronic Myelogenous Leukemia. Front. Oncol. 2019, 9, 435498. [Google Scholar] [CrossRef]
- Fonatsch, C.; Nowotny, H.; Pittermann-Höcker, E.; Streubel, B.; Jäger, U.; Valent, P.; Büchner, T.; Lechner, K. Amplification of Ribosomal RNA Genes in Acute Myeloid Leukemia. Genes Chromosom. Cancer 2001, 32, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Michaux, L.; Wlodarska, I.; Stul, M.; Dierlamm, J.; Mugneret, F.; Herens, C.; Beverloo, B.; Verhest, A.; Verellen-Dumoulin, C.; Verhoef, G.; et al. MLL Amplification in Myeloid Leukemias: A Study of 14 Cases with Multiple Copies of 11q23. Genes Chromosom. Cancer 2000, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Whittom, R.; Delage, R.; Drouin, R. A Unique Clone Involving Multiple Structural Chromosome Rearrangements in a Myelodysplastic Syndrome Case. Cancer Genet. Cytogenet. 2003, 140, 138–144. [Google Scholar] [CrossRef]
- Streubel, B.; Valent, P.; Lechner, K.; Fonatsch, C. Amplification of the AML1(CBFA2) Gene on Ring Chromosomes in a Patient with Acute Myeloid Leukemia and a Constitutional Ring Chromosome 21. Cancer Genet. Cytogenet. 2001, 124, 42–46. [Google Scholar] [CrossRef]
- Kakazu, N.; Taniwaki, M.; Horiike, S.; Nishida, K.; Tatekawa, T.; Nagai, M.; Takahashi, T.; Akaogi, T.; Inazawa, J.; Ohki, M.; et al. Combined Spectral Karyotyping and DAPI Banding Analysis of Chromosome Abnormalities in Myelodysplastic Syndrome. Genes Chromosom. Cancer 1999, 26, 336–345. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); S. Karger AG: Basel, Switzerland, 2020; ISBN 978-3-318-06867-2. [Google Scholar]
- Jiang, L.; Pallavajjala, A.; Huang, J.; Haley, L.; Morsberger, L.; Stinnett, V.; Hardy, M.; Park, R.; Ament, C.; Finch, A.; et al. Clinical Utility of Targeted Next-Generation Sequencing Assay to Detect Copy Number Variants Associated with Myelodysplastic Syndrome in Myeloid Malignancies. J. Mol. Diagn. 2021, 23, 467–483. [Google Scholar] [CrossRef]
- Toribio-Castelló, S.; Castaño, S.; Villaverde-Ramiro, Á.; Such, E.; Arnán, M.; Solé, F.; Díaz-Beyá, M.; Díez-Campelo, M.; del Rey, M.; González, T.; et al. Mutational Profile Enables the Identification of a High-Risk Subgroup in Myelodysplastic Syndromes with Isolated Trisomy 8. Cancers 2023, 15, 3822. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A Mutations in Patients with Myelodysplastic Syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Saitoh, S.; Lmoto, S.; Itoh, M.; Tsutsumi, M.; Hikiji, K.; Nakamura, H.; Matozaki, S.; Ogawa, R.; Nakao, Y.; et al. Multiple Point Mutation of N-Ras and K-Ras Oncogenes in Myelodysplastic Syndrome and Acute Myelogenous Leukemia. Oncology 1992, 49, 114–122. [Google Scholar] [CrossRef]
- Fuchs, O.; Provaznikova, D.; Kocova, M.; Kostecka, A.; Cvekova, P.; Neuwirtova, R.; Kobylka, P.; Cermak, J.; Brezinova, J.; Schwarz, J.; et al. CEBPA Polymorphisms and Mutations in Patients with Acute Myeloid Leukemia, Myelodysplastic Syndrome, Multiple Myeloma and Non-Hodgkin’s Lymphoma. Blood Cells Mol. Dis. 2008, 40, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.C.; Agosto-Peña, J. Epigenetic Regulation by ASXL1 in Myeloid Malignancies. Int. J. Hematol. 2023, 117, 791–806. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 Mutations in Primary Myelofibrosis: Clinical, Histopathology and Genetic Correlates among 155 Patients. Leukemia 2011, 26, 1135–1137. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why Are There Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas. Research Network Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Hou, H.A.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; Liu, C.W.; Tang, J.L.; et al. TP53 Mutations in de Novo Acute Myeloid Leukemia Patients: Longitudinal Follow-Ups Show the Mutation Is Stable during Disease Evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef]
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical Implications of Subclonal TP53 Mutations in Acute Myeloid Leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef]
- Invernizzi, R.; Filocco, A. Myelodysplastic Syndrome: Classification and Prognostic Systems. Oncol. Rev. 2010, 4, 25–33. [Google Scholar] [CrossRef]
- Mrózek, K.; Heinonen, K.; Theil, K.S.; Bloomfield, C.D. Spectral Karyotyping in Patients with Acute Myeloid Leukemia and a Complex Karyotype Shows Hidden Aberrations, Including Recurrent Overrepresentation of 21 q, 11q, and 22q. Genes Chromosom. Cancer 2002, 34, 137–153. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Pozdnyakova, O.; Geyer, J.T.; Cin, P.D.; Hasserjian, R.P. Ring Chromosome in Myeloid Neoplasms Is Associated with Complex Karyotype and Disease Progression. Hum. Pathol. 2017, 68, 40–46. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 Alterations in Acute Myeloid Leukemia with Complex Karyotype Correlate with Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis Is Linked to TP53 Alteration, Cell Cycle Impairment, and Dismal Outcome in Acute Myeloid Leukemia with Complex Karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef]
- Bochtler, T.; Granzow, M.; Stölzel, F.; Kunz, C.; Mohr, B.; Kartal-Kaess, M.; Hinderhofer, K.; Heilig, C.E.; Kramer, M.; Thiede, C.; et al. Marker Chromosomes Can Arise from Chromothripsis and Predict Adverse Prognosis in Acute Myeloid Leukemia. Blood 2017, 129, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Kurtas, N.E.; Xumerle, L.; Leonardelli, L.; Delledonne, M.; Brusco, A.; Chrzanowska, K.; Schinzel, A.; Larizza, D.; Guerneri, S.; Natacci, F.; et al. Small Supernumerary Marker Chromosomes: A Legacy of Trisomy Rescue? Hum. Mutat. 2019, 40, 193–200. [Google Scholar] [CrossRef]
- Liehr, T. Chapter 4—Formation of CG-CNVs. In Benign & Pathological Chromosomal Imbalances; Academic Press: Cambridge, MA, USA, 2014; Volume 1, pp. 29–36. ISBN 978-0-12-404631-3. [Google Scholar]
- Breman, A.; Stankiewicz, P. Chapter 2—Karyotyping as the First Genomic Approach. In Genomics of Rare Diseases: Understanding Disease Genetics Using Genomic Approaches; Academic Press: Cambridge, MA, USA, 2021; pp. 17–34. ISBN 9780128201404. [Google Scholar]
- Gisselsson, D.; Pettersson, L.; Höglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal Breakage-Fusion-Bridge Events Cause Genetic Intratumor Heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.C.; Tan, X.M.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor Suppressor TET2 Promotes Cancer Immunity and Immunotherapy Efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Kafer, G.R.; Li, X.; Horii, T.; Suetake, I.; Tajima, S.; Hatada, I.; Carlton, P.M. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep. 2016, 14, 1283–1292. [Google Scholar] [CrossRef]
- Kosztolányi, G. Does “Ring Syndrome” Exist? An Analysis of 207 Case Reports on Patients with a Ring Autosome. Hum. Genet. 1987, 75, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Kosztolányi, G.; Pap, M. Severe Growth Failure Associated with Atrophic Intestinal Mucosa and Ring Chromosome 15. Acta Paediatr. 1986, 75, 326–331. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Classification | Total Patients (%) | Mean Age at Diagnosis (Age Ranges) |
|---|---|---|
| Myeloid malignancies | ||
| Acute myeloid leukemia (AML) | 47 (48.0) | 67 (44–88) |
| Myelodysplastic syndromes (MDS) | 37 (37.8) | 70 (34–86) |
| Chronic myelomonocytic leukemia (CMML) → AML | 3 (3.1) | 62 (42–76) |
| Chronic myeloid leukemia (CML) → AML | 2 (2.0) | 60 (56–66) |
| CML | 1 (1.0) | 61 |
| Lymphoid malignancies | ||
| Multiple myeloma (MM) | 5 (5.1) | 61 (42–74) |
| Acute lymphoblastic leukemia (ALL) | 2 (2.0) | 40 (6–74) |
| Chronic lymphocytic leukemia (CLL) | 1 (1.0) | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyd, R.J.; Murry, J.B.; Morsberger, L.A.; Klausner, M.; Chen, S.; Gocke, C.D.; McCallion, A.S.; Zou, Y.S. Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants. Cancers 2023, 15, 5439. https://doi.org/10.3390/cancers15225439
Boyd RJ, Murry JB, Morsberger LA, Klausner M, Chen S, Gocke CD, McCallion AS, Zou YS. Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants. Cancers. 2023; 15(22):5439. https://doi.org/10.3390/cancers15225439
Chicago/Turabian StyleBoyd, Rachel J., Jaclyn B. Murry, Laura A. Morsberger, Melanie Klausner, Suping Chen, Christopher D. Gocke, Andrew S. McCallion, and Ying S. Zou. 2023. "Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants" Cancers 15, no. 22: 5439. https://doi.org/10.3390/cancers15225439
APA StyleBoyd, R. J., Murry, J. B., Morsberger, L. A., Klausner, M., Chen, S., Gocke, C. D., McCallion, A. S., & Zou, Y. S. (2023). Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants. Cancers, 15(22), 5439. https://doi.org/10.3390/cancers15225439