
Oligonucleotide-Based Therapeutics for STAT3 Targeting in Cancer—Drug Carriers Matter
 ,
, 
Abstract
:Simple Summary
Abstract
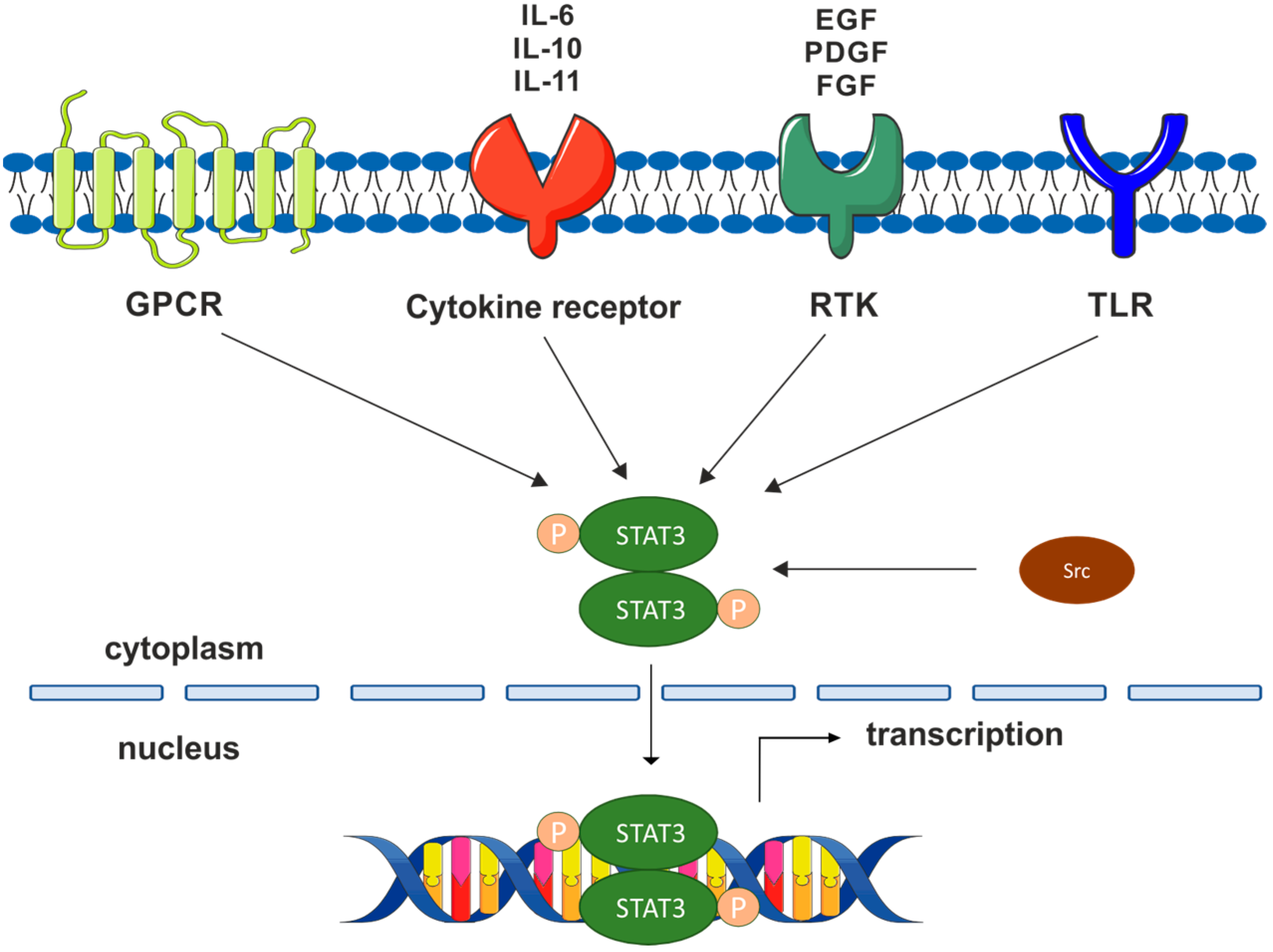
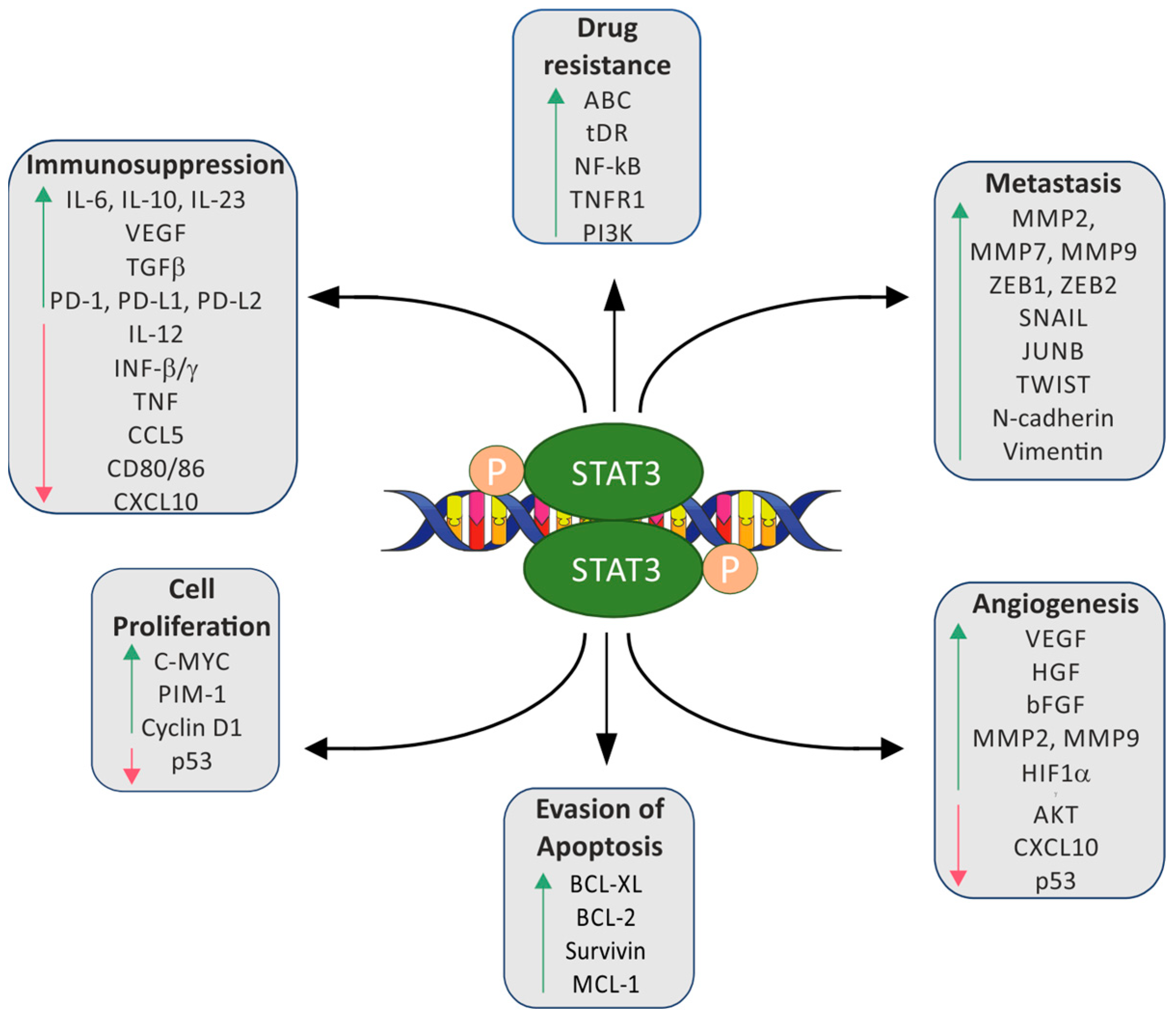
1. Introduction
2. Therapeutics Based on Nucleic Acids
2.1. Molecules of RNAi Pathway
Artificial RNAi Molecules
2.2. Antisense Oligonucleotides (ASO)
2.3. Oligodeoxynucleotide Decoys (ODN-Decoy)
3. Delivery of Therapeutics Based on Nucleic Acids
3.1. Chemical Modification of Oligonucleotide Therapeutics
3.2. Carriers Used for the Transportation of Oligonucleotide Therapeutics
4. Oligonucleotide-Based Therapeutics Targeting STAT3 Delivered into Cancer Cells in a Naked Form
4.1. siRNA-Based Therapeutics
4.2. shRNA-Based Therapeutics
4.3. ASO-Based Therapeutics
4.4. ODN-Decoy-Based Therapeutics

{kind=link}
{kind=link}
{kind=link}
| Oligo | Cancer | In Vitro Study | In Vivo Study | Ref. | ||
|---|---|---|---|---|---|---|
| Cell Line | Effect | Cell Line/Route of Administration | Effect | |||
| siRNA | Colorectal cancer | HCT-116 SW480 | Apoptosis ↑ | HCT-116 /intratumoral injection and electroporation | Tumor growth ↓ | [135] |
| Gastric cancer | SGC-7901 | Cell cycle arrest | SGC-7901 /local electrotransfection | Tumor growth ↓, proliferation ↓ | [136] | |
| Hepatocarcinoma | Bel-7402 | Apoptosis ↑ | nd | nd | [138] | |
| Lung cancer | Lewis lung cancer cell | Proliferation ↓ Apoptosis↑ | nd | nd | [139] | |
| HCC827 HCC827ER H1975 | Apoptosis ↑ | nd | nd | [140] | ||
| Leukemia | K562 | Proliferation ↓ Apoptosis ↑ Cell cycle arrest | nd | nd | [141] | |
| SU-DHL-8 OCI-LY1 | Proliferation ↓ Migration and invasion ↓ | nd | nd | [142] | ||
| nd | nd | MV4-11/intratumoral injection | Tumor growth ↓ | [143] | ||
| Myeloma | nd | nd | KMS-11/intratumoral injection | Tumor growth ↓ | [143] | |
| Ovarian cancer | SKOV3 OVCAR3 | Proliferation ↓ Apoptosis ↑ | OVCAR3 /in vitro modified cells | Tumor growth ↓ | [144] | |
| C13K SKOV3 | Chemoresistance ↓ | nd | nd | [145] | ||
| Astrocytoma | A172 T98G | Viability ↓ Apoptosis ↑ | nd | nd | [146] | |
| Retinoblastoma | ARPE-19 HRMECs Y79 | Proliferation ↓ | Y79 /intravitreal injection | Tumor growth ↓ | [147] | |
| Oral squamous cell carcinoma | GFP-SAS HSC-4 HSC-3 KB | Proliferation ↓ | nd | nd | [148] | |
| Breast cancer | MDA-MB-231 | Apoptosis ↑ | MDA-MB-231 /in vitro modified cells | Tumor growth ↓ Apoptosis ↑ | [151] | |
| MDA-MB-231 | Proliferation ↓ Migration ↓ Clonogenic survival ↓ | nd | nd | [152] | ||
| MDA-MB-231 HCC70 | Invasion ↓ | nd | nd | [82] | ||
| Prostate cancer | DU145 LN-17 | Proliferation ↓ Apoptosis ↑ | nd | nd | [153] | |
| nd | nd | PC3 DU145 /intratumoral injection | Tumor growth ↓ | [154] | ||
| shRNA | Ovarian cancer | CAOV3 | Proliferation ↓ Apoptosis ↑ | CAOV3/intratumoral injection /in vitro modified cells | Tumor growth ↓ Metastasis ↓ | [155,156] |
| Lung cancer | A549 SPC-A1 SK-MES-1 | Viability ↓ Colony formation ↓ Chemoresistance ↓ | A549 SK-MES-1 /in vitro modified cells | Radiosensitivity ↑ | [157,158] | |
| Oral cancer | nd | nd | Hep-2 /intratumoral injection | Radiosensitivity ↑ | [159] | |
| Head and neck cancer | HN5 | Proliferation ↓ | HN5 /in vitro modified cells | Tumor growth ↓ | [160] | |
| Prostate cancer | PC-3M | Apoptosis ↑ | PC-3M /intratumoral injection | Tumor growth ↓ | [161] | |
| Breast cancer | MCF7-HER2 | Invasiveness ↓ Tumorsphere formation ↓ Stemness ↓ | nd | nd | [162] | |
| Hepatocellular carcinoma | HepG2.2.15 | Proliferation ↓ Apoptosis ↑ | HepG2.2.15/intratumoral injection | Tumor growth ↓ | [163] | |
| Lymphoma | SCC-3 | Proliferation ↓ | SCC-3 /in vitro modified cells | Tumor growth ↓ | [164] | |
| ASO | Hepatocellular carcinoma | HCCLM3 SNU423 Huh7 | Proliferation ↓ Migration ↓ Apoptosis ↑ | HCCLM3/intraperitoneal injection | Angiogenesis ↓ Metastasis ↓ | [165] |
| Lymphoma | SUP-M2 KARPAS299 | Proliferation ↓ Apoptosis ↑ | A431 SUP-M2 /intravenous injection | Tumor growth ↓ | [89] | |
| ODN-decoy | Lung cancer | 201T H1975 | Apoptosis ↑ Colony formation ↓ | 201T H1975 /intravenous injection | Tumor growth ↓ | [168] |
| Breast cancer | MDA-MB-231 | Proliferation ↓ Mammospheroid formation ↓ Apoptosis ↑ Chemoresistance ↓ Radiosensitivity ↑ Invasion ↓ Migration ↓ | nd | nd | [92,95] | |
| Ovarian cancer | SKOV3 OVCAR3 | Chemoresistance ↓ Proliferation ↓ Apoptosis ↑ | SKOV3/intratumoral injection | Tumor growth ↓ Apoptosis ↑ | [169,170,171] | |
| Glioma | U251 A172 | Proliferation ↓ Apoptosis ↑ | nd U251 /intratumoral injection | nd Tumor growth ↓ Apoptosis ↑ | [172,173] | |
| Hepatocellular carcinoma | HepG2 PLC/PRF/5 H7402 | Apoptosis ↑ | nd | nd | [174] | |
| Colon cancer | SW480 | Chemoresistance ↓ Migration ↓ | nd | nd | [175] | |
| Head and neck cancer | nd | nd | UM-SCC1/intravenous injection | Tumor growth↓ | [176] | |
5. shRNA-Based Therapeutics Targeting STAT3 Delivered into Cancer Cells by Viral-Based Carrier Systems
6. Oligonucleotide-Based Therapeutics Targeting STAT3 Delivered into Cancer Cells by Non-Viral-Based Carrier Systems
6.1. siRNA-Based Therapeutics
6.2. shRNA-Based Therapeutics
6.3. ODN-Decoy-Based Therapeutics
| Oligo | Cancer | In Vitro Study | In Vivo Study | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Carrier | Cell Line | Effect | Carrier | Cell Line /Route of Administration | Effect | |||
| siRNA | Lung cancer | PEI/PLGA nanoparticles | A549 | Apoptosis ↑ Cell cycle arrest | PEI/PLGA nanoparticles | A549/intraperitoneal cavity injection | Apoptosis ↑ Cell cycle arrest | [186] |
| Ovarian cancer | PLGA/CSO micelles | SKOV3 | Proliferation ↓ Apoptosis ↑ | nd | nd | nd | [187] | |
| Colon cancer | rrPPC nanoparticles | C26 | Proliferation ↓ Apoptosis ↑ | rrPPC nanoparticles | C26/intravenous injection | Metastasis ↓ Tumor growth↓ Apoptosis ↑ Angiogenesis ↓ | [188] | |
| Colon cancer and breast cancer | TAT-FA-CLP nanoparticles | CT26 4T1 | Chemoresistance ↓ Apoptosis ↑ | TAT-FA-CLP nanoparticles | CT26 4T1 /tail vein injection | Proliferation ↓ Migration ↓ Invasion ↓ | [190] | |
| Breast cancer | 4T1 | Angiogenesis↓ Migration ↓ | Liposomes | 4T1 /tail vein injection | Metastasis ↓ Tumor growth↓ Apoptosis ↑ Angiogenesis ↓ | [192] | ||
| PR39 complexes | 4T1 | Migration ↓ Invasion ↓ | nd | nd | nd | [193] | ||
| HA/PPL micelles | 4T1 | Chemoresistance↓ | HA/PPL micelles | 4T1 /intravenous injection | Metastasis ↓ Tumor growth↓ | [194] | ||
| chMSNs | MCF7 | Chemoresistance↓ | nd | nd | nd | [195] | ||
| nd | nd | nd | PLL-PEG nanoparticles | 4T1 | Tumor growth↓ | [196] | ||
| Melanoma | nd | nd | nd | Cationic liposomes | B16F10/intratumoral injection | Tumor growth ↓ | [197] | |
| Cationic curdlan nanoparticles | B16 | Apoptosis ↑ | nd | nd | nd | [198] | ||
| PEI polyplexes | B16.F10 | Cell viability ↓ | nd | nd | nd | [124] | ||
| PEI complexes + microneedles | B16.F10 | Proliferation ↓ | PEI complexes + microneedles | B16.F10/intratumoral injection | Tumor growth↓ | [199] | ||
| shRNA | Ovarian cancer | nd | A2780CPA2780s | Proliferation ↓ Apoptosis ↑ | Cationic liposomes | A2780CP/intraperitoneal cavity injection | Tumor growth↓ Apoptosis ↑ | [200] |
| Lung cancer | nd | H1650 | Proliferation ↓ Apoptosis ↑ | VCH nanoparticles | H1650 /in vitro modified cells | Tumor growth↓ | [201] | |
| ODN-decoy | Esophageal squamous cell carcinoma | Ultrasound SonoVue microbubbles + irradiation | EC9706 | Proliferation ↓ | Ultrasound SonoVue microbubbles + irradiation | EC9706 /intravenous injection | Tumor growth↓ | [202] |
| Ovarian cancer | nd | nd | nd | SLN nanoparticles | SKOV3/intratumoral injection | Tumor growth↓ Apoptosis ↑ | [203] | |
| Breast cancer | CaP@HA nanoparticles | BT474R | Chemoresistance↓ Cell viability ↓ Apoptosis ↑ | CaP@HA nanoparticles | BT474R/intravenous injection | Tumor growth↓ Apoptosis ↑ Survival ↑ | [204] | |
| Head and neck cancer | AuNP-NUAP nanoparticles | FaDu | Radiosensitivity ↑ Proliferation ↓ | nd | nd | nd | [133] | |
7. Oligonucleotide-Based Therapeutics Targeting STAT3 in Cancer Investigated in the Clinic
7.1. siRNA-Based Therapeutics
7.2. ASO-Based Therapeutics
7.3. ODN-Decoy-Based Therapeutics
8. Conclusions—Drug Carriers Matter
9. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Darnell, J.E.; Kerr Ian, M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Shriki, A.; Galun, E.; Axelrod, J.H.; Daum, H.; Rottenberg, Y.; Hamburger, T.; Mali, B.; Peretz, T. Tissue Microarray-Based Study of Patients with Lymph Node-Positive Breast Cancer Shows Tyrosine Phosphorylation of Signal Transducer and Activator of Transcription 3 (Tyrosine705-STAT3) Is a Marker of Good Prognosis. Clin. Transl. Oncol. 2012, 14, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 Pathway as a Therapeutic Target in Head and Neck Cancer: Barriers and Innovations. Oral. Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Saini, U.; Naidu, S.; ElNaggar, A.C.; Bid, H.K.; Wallbillich, J.J.; Bixel, K.; Bolyard, C.; Suarez, A.A.; Kaur, B.; Kuppusamy, P.; et al. Elevated STAT3 Expression in Ovarian Cancer Ascites Promotes Invasion and Metastasis: A Potential Therapeutic Target. Oncogene 2017, 36, 168–181. [Google Scholar] [CrossRef] [PubMed]
- To, S.Q.; Dmello, R.S.; Richards, A.K.; Ernst, M.; Chand, A.L. STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers 2022, 14, 429. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Giraud, A.S.; Menheniott, T.R.; Judd, L.M. Targeting STAT3 in Gastric Cancer. Expert. Opin. Ther. Targets 2012, 16, 889–901. [Google Scholar] [CrossRef]
- Bishop, J.; Thaper, D.; Zoubeidi, A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers 2014, 6, 829–859. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Targeting the STAT3 Signaling Pathway in Cancer: Role of Synthetic and Natural Inhibitors. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1845, 136–154. [Google Scholar] [CrossRef]
- Mitchell, T.J.; John, S. Signal Transducer and Activator of Transcription (STAT) Signalling and T-Cell Lymphomas. Immunology 2005, 114, 301–312. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but Not Stat1 Activation for Epidermal Growth Factor Receptor-Mediated Cell Growth In Vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Müller-Newen, G.; Küster, A.; Wijdenes, J.; Schaper, F.; Heinrich, P.C. Studies on the Interleukin-6-Type Cytokine Signal Transducer Gp130 Reveal a Novel Mechanism of Receptor Activation by Monoclonal Antibodies. J. Biol. Chem. 2000, 275, 4579–4586. [Google Scholar] [CrossRef]
- Guschin, D.; Rogers, N.; Briscoe, J.; Witthuhn, B.; Watling, D.; Horn, F.; Pellegrini, S.; Yasukawa, K.; Heinrich, P.; Stark, G.R. A Major Role for the Protein Tyrosine Kinase JAK1 in the JAK/STAT Signal Transduction Pathway in Response to Interleukin-6. EMBO J. 1995, 14, 1421–1429. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Lin, L.; He, L.; Wu, Y.; Zhang, L.; Yi, Z.; Chen, Y.; Pang, X.; Liu, M. Inhibition of STAT3 Signaling Pathway by Nitidine Chloride Suppressed the Angiogenesis and Growth of Human Gastric Cancer. Mol. Cancer Ther. 2012, 11, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in Oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Wu, E.H.T.; Lo, R.K.H.; Wong, Y.H. Regulation of STAT3 Activity by G16-Coupled Receptors. Biochem. Biophys. Res. Commun. 2003, 303, 920–925. [Google Scholar] [CrossRef]
- Liu, B.-S.; Cao, Y.; Huizinga, T.W.; Hafler, D.A.; Toes, R.E.M. TLR-Mediated STAT3 and ERK Activation Controls IL-10 Secretion by Human B Cells. Eur. J. Immunol. 2014, 44, 2121–2129. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal Activation of Transcription by Statl and Stat3 Requires Both Tyrosine and Serine Phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 Sequence-Specific DNA Binding and Transcription by P300/CREB-Binding Protein-Mediated Acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Guan, Y.; Chatterjee, D.; Chin, Y.E. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Man, Q.; Huo, F.; Gao, X.; Lin, H.; Li, S.; Wang, J.; Su, F.; Cai, L.; Shi, Y.; et al. STAT3 Pathway in Cancers: Past, Present, and Future. MedComm 2022, 3, e124. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Mohammad, I.; Liu, Z. Overview of the STAT-3 Signaling Pathway in Cancer and the Development of Specific Inhibitors (Review). Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-Mediated Myc Expression Is Required for Src Transformation and PDGF-Induced Mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S.; et al. Gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yan, R.; He, X.; He, J. Constitutive Activation of STAT3 and Cyclin D1 Overexpression Contribute to Proliferation, Migration and Invasion in Gastric Cancer Cells. Am. J. Transl. Res. 2017, 9, 5671–5677. [Google Scholar]
- Deepak Roshan, V.G.; Sinto, M.S.; Thomas, S.; Kannan, S. Cyclin D1 Overexpression Associated with Activation of STAT3 in Oral Carcinoma Patients from South India. J. Cancer Res. Ther. 2018, 14, 403–408. [Google Scholar] [CrossRef]
- Shirogane, T.; Fukada, T.; Muller, J.M.M.; Shima, D.T.; Hibi, M.; Hirano, T. Synergistic Roles for Pim-1 and c-Myc in STAT3-Mediated Cell Cycle Progression and Antiapoptosis. Immunity 1999, 11, 709–719. [Google Scholar] [CrossRef]
- Pensa, S.; Regis, G.; Boselli, D.; Novelli, F.; Poli, V. STAT1 and STAT3 in Tumorigenesis: Two Sides of the Same Coin? Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Yu, H.; Jove, R. The STATs of Cancer—New Molecular Targets Come of Age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zhang, Z.-G.; Tian, X.-Q.; Sun, D.-F.; Liang, Q.-C.; Zhang, Y.-J.; Lu, R.; Chen, Y.-X.; Fang, J.-Y. Inhibition of JAK1, 2/STAT3 Signaling Induces Apoptosis, Cell Cycle Arrest, and Reduces Tumor Cell Invasion in Colorectal Cancer Cells. Neoplasia 2008, 10, 287–297. [Google Scholar] [CrossRef]
- Jiao, Y.; Ding, H.; Huang, S.; Liu, Y.; Sun, X.; Wei, W.; Ma, J.; Zheng, F. Bcl-XL and Mcl-1 Upregulation by Calreticulin Promotes Apoptosis Resistance of Fibroblast-like Synoviocytes via Activation of PI3K/Akt and STAT3 Pathways in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2018, 36, 841–849. [Google Scholar]
- Kanda, N.; Seno, H.; Konda, Y.; Marusawa, H.; Kanai, M.; Nakajima, T.; Kawashima, T.; Nanakin, A.; Sawabu, T.; Uenoyama, Y.; et al. STAT3 Is Constitutively Activated and Supports Cell Survival in Association with Survivin Expression in Gastric Cancer Cells. Oncogene 2004, 23, 4921–4929. [Google Scholar] [CrossRef] [PubMed]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 Regulates Genes Common to Both Wound Healing and Cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Suiqing, C.; Min, Z.; Lirong, C. Overexpression of Phosphorylated-STAT3 Correlated with the Invasion and Metastasis of Cutaneous Squamous Cell Carcinoma. J. Dermatol. 2005, 32, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively Activated Stat3 Induces Tumorigenesis and Enhances Cell Motility of Prostate Epithelial Cells through Integrin Β6. Mol. Cell Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef]
- Gao, J.; McConnell, M.J.; Yu, B.; Li, J.; Balko, J.M.; Black, E.P.; Johnson, J.O.; Lloyd, M.C.; Altiok, S.; Haura, E.B. MUC1 Is a Downstream Target of STAT3 and Regulates Lung Cancer Cell Survival and Invasion. Int. J. Oncol. 2009, 35, 337–345. [Google Scholar]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated Signal Transducer and Activator of Transcription (STAT) 3: Localization in Focal Adhesions and Function in Ovarian Cancer Cell Motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Rajasekhar Maddirela, D.; Gujrati, M.; Rao, J.S. Essential Role of Cooperative NF-κB and Stat3 Recruitment to ICAM-1 Intronic Consensus Elements in the Regulation of Radiation-Induced Invasion and Migration in Glioma. Oncogene 2013, 32, 5144–5155. [Google Scholar] [CrossRef]
- Ng, D.C.H.; Lin, B.H.; Lim, C.P.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 Regulates Microtubules by Antagonizing the Depolymerization Activity of Stathmin. J. Cell Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Pandya, P.; Dalmeida, R.; Karagiannis, P.; Sanchez-Laorden, B.; Viros, A.; Albrengues, J.; Nestle, F.O.; Ridley, A.J.; Gaggioli, C.; et al. Diverse Matrix Metalloproteinase Functions Regulate Cancer Amoeboid Migration. Nat. Commun. 2014, 5, 4255. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sabri, N.; Li, J.; Li, W.X. Role of STAT3 in Lung Cancer. JAK-STAT 2014, 3, e999503. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 Activation Regulates the Expression of Matrix Metalloproteinase-2 and Tumor Invasion and Metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Qian, L.; Song, S.; Chen, L.; Zhang, Y.; Yuan, G.; Zhang, H.; Xia, Q.; Hu, M.; Yu, M.; et al. Fra-1 and Stat3 Synergistically Regulate Activation of Human MMP-9 Gene. Mol. Immunol. 2008, 45, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Tsareva, A.S.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schutz, A.; Kovacic, B.; Friedrich, K. Signal Transducer and Activator of Transcription 3 Activation Promotes Invasive Growth of Colon Carcinomas through Matrix Metal Loproteinase Induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Zhang, W.; Sun, M.; Wang, Q.; Coppola, D.; Mansour, M.; Xu, L.; Costanzo, C.; Cheng, J.Q.; Wang, L.-H. Twist Is Transcriptionally Induced by Activation of STAT3 and Mediates STAT3 Oncogenic Function. J. Biol. Chem. 2008, 283, 14665–14673. [Google Scholar] [CrossRef]
- Alexander, N.R.; Tran, N.L.; Rekapally, H.; Summers, C.E.; Glackin, C.; Heimark, R.L. N-Cadherin Gene Expression in Prostate Carcinoma Is Modulated by Integrin-Dependent Nuclear Translocation of Twist1. Cancer Res. 2006, 66, 3365–3369. [Google Scholar] [CrossRef]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist Is a Transcriptional Repressor of E-Cadherin Gene Expression in Breast Cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef]
- Liu, W.-H.; Chen, M.-T.; Wang, M.-L.; Lee, Y.-Y.; Chiou, G.-Y.; Chien, C.-S.; Huang, P.-I.; Chen, Y.-W.; Huang, M.-C.; Chiou, S.-H.; et al. Cisplatin-Selected Resistance Is Associated with Increased Motility and Stem-like Properties via Activation of STAT3/Snail Axis in Atypical Teratoid/Rhabdoid Tumor Cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 Pathway in the Conversion of Non-Stem Cancer Cells into Cancer Stem-like Cells. Cell Signal 2013, 25, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Potent Synergism between Vascular Endothelial Growth Factor and Basic Fibroblast Growth Factor in the Induction of Angiogenesis in Vitro. Biochem. Biophys. Res. Commun. 1992, 189, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 Activity Up-Regulates VEGF Expression and Tumor Angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Chou, C.-H.; Lai, K.-B.; Lee, C.-N.; Hsieh, C.-Y. Interleukin-6 Promotes Cervical Tumor Growth by VEGF-Dependent Angiogenesis via a STAT3 Pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 Activation Regulates the Expression of Vascular Endothelial Growth Factor and Human Pancreatic Cancer Angiogenesis and Metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-C.; Liu, X.; Li, M.-M.; Ma, Y.-Y.; Sun, H.-T.; Tian, X.-Y.; Wang, Y.; Liu, M.; Fu, L.-S.; Wang, Y.-F.; et al. AT-533, a Novel Hsp90 Inhibitor, Inhibits Breast Cancer Growth and HIF-1α/VEGF/VEGFR-2-Mediated Angiogenesis in Vitro and in Vivo. Biochem. Pharmacol. 2020, 172, 113771. [Google Scholar] [CrossRef]
- Bi, Y.-H.; Han, W.-Q.; Li, R.-F.; Wang, Y.-J.; Du, Z.-S.; Wang, X.-J.; Jiang, Y. Signal Transducer and Activator of Transcription 3 Promotes the Warburg Effect Possibly by Inducing Pyruvate Kinase M2 Phosphorylation in Liver Precancerous Lesions. World J. Gastroenterol. 2019, 25, 1936–1949. [Google Scholar] [CrossRef]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.-L. STAT3 Inhibition, a Novel Approach to Enhancing Targeted Therapy in Human Cancers. Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar] [CrossRef]
- Demartis, A.; Bernassola, F.; Savino, R.; Melino, G.; Ciliberto, G. Interleukin 6 Receptor Superantagonists Are Potent Inducers of Human Multiple Myeloma Cell Death. Cancer Res. 1996, 56, 4213–4218. [Google Scholar]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Kong, Y.; Yu, Z.-B.; Bai, L.; Xiao, Y.-B. Hypoxia-Induced SOCS3 Is Limiting STAT3 Phosphorylation and NF-κB Activation in Congenital Heart Disease. Biochimie 2011, 93, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, C.; He, J.; Guo, Q.; Hu, D.; Yang, X.; Wang, J.; Kang, Y.; She, R.; Wang, Z.; et al. STAT3 Inhibitor Stattic Enhances Radiosensitivity in Esophageal Squamous Cell Carcinoma. Tumor Biol. 2015, 36, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A Low-Molecular-Weight Compound Discovered through Virtual Database Screening Inhibits Stat3 Function in Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yuan, X.; Shen, Y.-H.; Wang, J.; Azeem, W.; Yang, S.; Gade, A.; Lellahi, S.M.; Øyan, A.M.; Ke, X.; et al. Novel STAT3 Inhibitors Targeting STAT3 Dimerization by Binding to the STAT3 SH2 Domain. Front. Pharmacol. 2022, 13, 836724. [Google Scholar] [CrossRef]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, K.T.G.; Crews, C.M. Targeted Protein Degradation: A Promise for Undruggable Proteins. Cell Chem. Biol. 2021, 28, 934–951. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically Exploiting STAT3 Activity in Cancer—Using Tissue Repair as a Road Map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Zogg, H.; Singh, R.; Ro, S. Current Advances in RNA Therapeutics for Human Diseases. Int. J. Mol. Sci. 2022, 23, 2736. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther.-Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Alshaer, W.; Zureigat, H.; Al Karaki, A.; Al-Kadash, A.; Gharaibeh, L.; Hatmal, M.M.; Aljabali, A.A.A.; Awidi, A. siRNA: Mechanism of Action, Challenges, and Therapeutic Approaches. Eur. J. Pharmacol. 2021, 905, 174178. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Tomari, Y.; Zamore, P.D. Perspective: Machines for RNAi. Genes Dev. 2005, 19, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Traber, G.M.; Yu, A.-M. RNAi-Based Therapeutics and Novel RNA Bioengineering Technologies. J. Pharmacol. Exp. Ther. 2023, 384, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA Sponges: Competitive Inhibitors of Small RNAs in Mammalian Cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Kobayashi, H.; Singer, R.H. Single-Molecule Imaging of microRNA-Mediated Gene Silencing in Cells. Nat. Commun. 2022, 13, 1435. [Google Scholar] [CrossRef]
- Eisen, T.J.; Eichhorn, S.W.; Subtelny, A.O.; Bartel, D.P. MicroRNAs Cause Accelerated Decay of Short-Tailed Target mRNAs. Mol. Cell 2020, 77, 775–785.e8. [Google Scholar] [CrossRef]
- Svoboda, P. Key Mechanistic Principles and Considerations Concerning RNA Interference. Front. Plant Sci. 2020, 11, 1237. [Google Scholar] [CrossRef]
- McDaniel, J.M.; Varley, K.E.; Gertz, J.; Savic, D.S.; Roberts, B.S.; Bailey, S.K.; Shevde, L.A.; Ramaker, R.C.; Lasseigne, B.N.; Kirby, M.K.; et al. Genomic Regulation of Invasion by STAT3 in Triple Negative Breast Cancer. Oncotarget 2017, 8, 8226–8238. [Google Scholar] [CrossRef]
- Cardarelli, F.; Digiacomo, L.; Marchini, C.; Amici, A.; Salomone, F.; Fiume, G.; Rossetta, A.; Gratton, E.; Pozzi, D.; Caracciolo, G. The Intracellular Trafficking Mechanism of Lipofectamine-Based Transfection Reagents and Its Implication for Gene Delivery. Sci. Rep. 2016, 6, 25879. [Google Scholar] [CrossRef]
- Sheng, P.; Flood, K.A.; Xie, M. Short Hairpin RNAs for Strand-Specific Small Interfering RNA Production. Front. Bioeng. Biotechnol. 2020, 8, 940. [Google Scholar] [CrossRef]
- Taxman, D.J.; Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. In RNA Therapeutics; Sioud, M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 629, pp. 139–156. ISBN 978-1-60761-656-6. [Google Scholar]
- Xu, W.; Jiang, X.; Huang, L. RNA Interference Technology. In Comprehensive Biotechnology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 560–575. ISBN 978-0-444-64047-5. [Google Scholar]
- Bofill-De Ros, X.; Gu, S. Guidelines for the Optimal Design of miRNA-Based shRNAs. Methods 2016, 103, 157–166. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-Generation Antisense Oligonucleotide Inhibitor of STAT3 with Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Liang, X.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation Efficiency of mRNAs Is Increased by Antisense Oligonucleotides Targeting Upstream Open Reading Frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef]
- Rahmati, M.; Johari, B.; Kadivar, M.; Rismani, E.; Mortazavi, Y. Suppressing the Metastatic Properties of the Breast Cancer Cells Using STAT3 Decoy Oligodeoxynucleotides: A Promising Approach for Eradication of Cancer Cells by Differentiation Therapy. J. Cell Physiol. 2020, 235, 5429–5444. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Xu, X.; Shen, L.; Yao, Y.; Yang, Z.; Liu, P. STAT3 Decoy Oligodeoxynucleotides-Loaded Solid Lipid Nanoparticles Induce Cell Death and Inhibit Invasion in Ovarian Cancer Cells. PLoS ONE 2015, 10, e0124924. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Blenke, E.O.; Mastrobattista, E. Oligonucleotides. In Pharmaceutical Biotechnology; Crommelin, D.J.A., Sindelar, R.D., Meibohm, B., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 305–322. ISBN 978-3-030-00709-6. [Google Scholar]
- Johari, B.; Rahmati, M.; Nasehi, L.; Mortazavi, Y.; Faghfoori, M.H.; Rezaeejam, H. Evaluation of STAT3 Decoy Oligodeoxynucleotides’ Synergistic Effects on Radiation and/or Chemotherapy in Metastatic Breast Cancer Cell Line. Cell Biol. Int. 2020, 44, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, H.J.; Green, J.J.; Tzeng, S.Y. Cancer-Targeting Nanoparticles for Combinatorial Nucleic Acid Delivery. Adv. Mater. 2020, 32, 1901081. [Google Scholar] [CrossRef] [PubMed]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjörner, E.K.; Stevens, M.M.; et al. Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J.-L.; Li, P.; Rait, V.; Sergueeva, Z.A.; Sergueev, D. Reading, Writing, and Modulating Genetic Information with Boranophosphate Mimics of Nucleotides, DNA, and RNA. Ann. N. Y. Acad. Sci. 2003, 1002, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. Design of Antisense Oligonucleotides Stabilized by Locked Nucleic Acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Thonberg, H.; Ljungberg, K.; Frieden, M.; Westergaard, M.; Xu, Y.; Wahren, B.; Liang, Z.; Ørum, H.; Koch, T.; et al. Locked Nucleic Acid (LNA) Mediated Improvements in siRNA Stability and Functionality. Nucleic Acids Res. 2005, 33, 439–447. [Google Scholar] [CrossRef]
- Baker, Y.R.; Thorpe, C.; Chen, J.; Poller, L.M.; Cox, L.; Kumar, P.; Lim, W.F.; Lie, L.; McClorey, G.; Epple, S.; et al. An LNA-Amide Modification That Enhances the Cell Uptake and Activity of Phosphorothioate Exon-Skipping Oligonucleotides. Nat. Commun. 2022, 13, 4036. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Pakula, M.M.; Hansen, T.B.; Bus, C.; Langkjær, N.; Odadzic, D.; Smicius, R.; Wengel, S.L.; Chattopadhyaya, J.; Engels, J.W.; et al. A Screen of Chemical Modifications Identifies Position-Specific Modification by UNA to Most Potently Reduce siRNA off-Target Effects. Nucleic Acids Res. 2010, 38, 5761–5773. [Google Scholar] [CrossRef]
- Krichevsky, A.M.; Uhlmann, E.J. Oligonucleotide Therapeutics as a New Class of Drugs for Malignant Brain Tumors: Targeting mRNAs, Regulatory RNAs, Mutations, Combinations, and Beyond. Neurotherapeutics 2019, 16, 319–347. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Seong, B.L. Exploiting Virus-like Particles as Innovative Vaccines against Emerging Viral Infections. J. Microbiol. 2017, 55, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Qushawy, M.; Nasr, A. Solid Lipid Nanoparticles (Slns) As Nano Drug Delivery Carriers: Preparation, Characterization and Application. Int. J. Appl. Pharm. 2019, 12, 1–9. [Google Scholar] [CrossRef]
- Jeong, M.; Lee, Y.; Park, J.; Jung, H.; Lee, H. Lipid Nanoparticles (LNPs) for in Vivo RNA Delivery and Their Breakthrough Technology for Future Applications. Adv. Drug Deliv. Rev. 2023, 200, 114990. [Google Scholar] [CrossRef]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid Nanoparticles for Delivery of Therapeutic RNA Oligonucleotides. Mol. Pharm. 2019, 16, 2265–2277. [Google Scholar] [CrossRef]
- Belletti, D.; Tosi, G.; Forni, F.; Lagreca, I.; Barozzi, P.; Pederzoli, F.; Vandelli, M.A.; Riva, G.; Luppi, M.; Ruozi, B. PEGylated siRNA Lipoplexes for Silencing of BLIMP-1 in Primary Effusion Lymphoma: In Vitro Evidences of Antitumoral Activity. Eur. J. Pharm. Biopharm. 2016, 99, 7–17. [Google Scholar] [CrossRef]
- Li, Y.; Liu, R.; Yang, J.; Ma, G.; Zhang, Z.; Zhang, X. Dual Sensitive and Temporally Controlled Camptothecin Prodrug Liposomes Codelivery of siRNA for High Efficiency Tumor Therapy. Biomaterials 2014, 35, 9731–9745. [Google Scholar] [CrossRef]
- Khatri, N.; Baradia, D.; Vhora, I.; Rathi, M.; Misra, A. Development and Characterization of siRNA Lipoplexes: Effect of Different Lipids, In Vitro Evaluation in Cancerous Cell Lines and In Vivo Toxicity Study. AAPS PharmSciTech 2014, 15, 1630–1643. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and Persistent in Vivo Anti-HBV Activity of Chemically Modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Y.; Shen, S.; Zhu, Y.; Wang, J. Targeting Glucose Uptake of Glioma Cells by siRNA Delivery with Polymer Nanoparticle. J. Control. Release 2015, 213, e23–e24. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-Mediated Gene Silencing in Non-Human Primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Hattab, D.; Gazzali, A.M.; Bakhtiar, A. Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics 2021, 13, 1009. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and Safety of Patisiran for Familial Amyloidotic Polyneuropathy: A Phase II Multi-Dose Study. Orphanet J. Rare Dis. 2015, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Mendonça, S.L.; Matos, L.; Prata, M.J.; Jurado, S.A.; Pedroso de Lima, M.C.; Alves, S. Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach. Int. J. Mol. Sci. 2020, 21, 5732. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi Chalbatani, G.; Dana, H.; Gharagouzloo, E.; Grijalvo, S.; Eritja, R.; Logsdon, C.D.; Memari, F.; Miri, S.R.; Rezvani Rad, M.; Marmari, V. Small Interfering RNAs (siRNAs) in Cancer Therapy: A Nano-Based Approach. Int. J. Nanomed. 2019, 14, 3111–3128. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Jiang, K.; Zhang, Y.; Liu, Y.; Hu, G.; Song, J. Biosafety Assessment of Delivery Systems for Clinical Nucleic Acid Therapeutics. Biosaf. Health 2022, 4, 105–117. [Google Scholar] [CrossRef]
- Sun, B.; Wu, W.; Narasipura, E.A.; Ma, Y.; Yu, C.; Fenton, O.S.; Song, H. Engineering Nanoparticle Toolkits for mRNA Delivery. Adv. Drug Deliv. Rev. 2023, 200, 115042. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Nanomedicine: Current Status and Future Prospects. FASEB J. 2005, 19, 311–330. [Google Scholar] [CrossRef]
- Maghsoudnia, N.; Eftekhari, R.B.; Sohi, A.N.; Zamzami, A.; Dorkoosh, F.A. Application of Nano-Based Systems for Drug Delivery and Targeting: A Review. J. Nanopart. Res. 2020, 22, 245. [Google Scholar] [CrossRef]
- Alshamsan, A. STAT3-siRNA Induced B16.F10 Melanoma Cell Death: More Association with VEGF Downregulation than p-STAT3 Knockdown. Saudi Pharm. J. 2018, 26, 1083–1088. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug Delivery Systems for RNA Therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Nandgude, T.; Pagar, R. Plausible Role of Chitosan in Drug and Gene Delivery against Resistant Breast Cancer Cells. Carbohydr. Res. 2021, 506, 108357. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, C.-H.; Wayne Pack, D. Polymeric Carriers for Gene Delivery: Chitosan and Poly(Amidoamine) Dendrimers. CPD 2010, 16, 2350–2368. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Williams, G.R.; Wu, J.; Wu, J.; Zhang, X.; Zheng, H.; Li, S.; Zhu, L.-M. A Novel Chitosan-Based Nanomedicine for Multi-Drug Resistant Breast Cancer Therapy. Chem. Eng. J. 2019, 369, 134–149. [Google Scholar] [CrossRef]
- Su, W.P.; Cheng, F.Y.; Shieh, D.B.; Yeh, C.S.; Su, W.C. PLGA Nanoparticles Codeliver Paclitaxel and Stat3 siRNA to Overcome Cellular Resistance in Lung Cancer Cells. Int. J. Nanomed. 2012, 7, 4269–4283. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Cui, Y.; Zheng, L.; Kah-Hoe Chow, P.; Wang, C.-H. Development of a Gene/Drug Dual Delivery System for Brain Tumor Therapy: Potent Inhibition via RNA Interference and Synergistic Effects. Biomaterials 2013, 34, 7483–7494. [Google Scholar] [CrossRef] [PubMed]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef]
- Zhang, S.; Gupta, S.; Fitzgerald, T.J.; Bogdanov, A.A. Dual Radiosensitization and Anti-STAT3 Anti-Proliferative Strategy Based on Delivery of Gold Nanoparticle-Oligonucleotide Nanoconstructs to Head and Neck Cancer Cells. Nanotheranostics 2018, 2, 1–11. [Google Scholar] [CrossRef]
- Li, W.; Cao, Z.; Liu, R.; Liu, L.; Li, H.; Li, X.; Chen, Y.; Lu, C.; Liu, Y. AuNPs as an Important Inorganic Nanoparticle Applied in Drug Carrier Systems. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4222–4233. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Yang, X.; Shen, D.; Sun, H.; Huang, K.; Zheng, H. Effects and Mechanism of STAT3 Silencing on the Growth and Apoptosis of Colorectal Cancer Cells. Oncol. Lett. 2018, 16, 5575–5582. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, B.; Xu, L.; Zhong, J.; Liu, Z.; Liang, H.; Wen, N.; Yun, W.; Zhang, L.; Zhao, X. Stat3-siRNA Inhibits the Growth of Gastric Cancer in Vitro and in Vivo: Stat3-siRNA Inhibits Gastric Cancer Growth. Cell Biochem. Funct. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jin, R.; Wang, W.; Zhang, T.; Sang, J.; Li, N.; Han, Q.; Zhao, W.; Li, C.; Liu, Z. STAT3 Regulates Glycolysis via Targeting Hexokinase 2 in Hepatocellular Carcinoma Cells. Oncotarget 2017, 8, 24777–24784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, J.; Liu, Q.; Zhang, Y. Down-Regulation of STAT3 Expression Using Vector-Based RNA Interference Promotes Apoptosis in Hepatocarcinoma Cells. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1201–1205. [Google Scholar] [CrossRef]
- Wang, C.; Sun, M.; Zhao, X.; Zhang, X. Effect of STAT3 siRNA-Induced Inhibition of STAT3 Gene Expression on the Growth and Apoptosis of Lewis Lung Cancer Cells. Chin. J. Clin. Oncol. 2006, 3, 392–399. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, B.; Ning, Q.; Xia, Z.; Zhong, R.; Zhang, L.; Wu, L. Combination of Huanglian Jiedu Decoction and Erlotinib Delays Growth and Improves Sensitivity of EGFR-mutated NSCLC Cells in Vitro and in Vivo via STAT3/Bcl-2 Signaling. Oncol. Rep. 2020, 45, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhou, M.; Wen, S.; Ni, C.; Jiang, L.; Fan, J.; Xia, L. Effects of STAT3 Silencing on Fate of Chronic Myelogenous Leukemia K562 Cells. Leuk. Lymphoma 2010, 51, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Fan, C.; Ma, J.; Xue, H.; Xu, L. STAT3 Promotes Cell Proliferation by Potentiating the CCL4 Transcriptional Activity in Diffuse Large B-Cell Lymphoma. Acta Haematol. 2021, 145, 371–383. [Google Scholar] [CrossRef]
- Zhang, Q.; Hossain, D.M.S.; Nechaev, S.; Kozlowska, A.; Zhang, W.; Liu, Y.; Kowolik, C.M.; Swiderski, P.; Rossi, J.J.; Forman, S.; et al. TLR9-Mediated siRNA Delivery for Targeting of Normal and Malignant Human Hematopoietic Cells in Vivo. Blood 2013, 121, 1304–1315. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, G.; Tong, X.; You, Q.; An, Y.; Wang, Y.; Guo, L.; Wang, T.; Zhu, D.; Zheng, J. Growth Inhibition of Human Ovarian Cancer Cells by Blocking STAT3 Activation with Small Interfering RNA. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 148, 73–80. [Google Scholar] [CrossRef]
- Han, Z.; Feng, J.; Hong, Z.; Chen, L.; Li, W.; Liao, S.; Wang, X.; Ji, T.; Wang, S.; Ma, D.; et al. Silencing of the STAT3 Signaling Pathway Reverses the Inherent and Induced Chemoresistance of Human Ovarian Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 435, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Konnikova, L.; Kotecki, M.; Kruger, M.M.; Cochran, B.H. Knockdown of STAT3 Expression by RNAi Induces Apoptosis in Astrocytoma Cells. BMC Cancer 2003, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.H.; Kim, J.H.; Cho, C.S.; Cho, Y.-L.; Jun, H.O.; Yu, Y.S.; Min, J.-K.; Kim, J.H. STAT3 Inhibition Suppresses Proliferation of Retinoblastoma through Down-Regulation of Positive Feedback Loop of STAT3/miR-17-92 Clusters. Oncotarget 2014, 5, 11513–11525. [Google Scholar] [CrossRef] [PubMed]
- Klosek, S.K.; Nakashiro, K.I.; Hara, S.; Goda, H.; Hamakawa, H. Stat3 as a Molecular Target in RNA Interference-Based Treatment of Oral Squamous Cell Carcinoma. Oncol. Rep. 2008, 20, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Lu, X.; Di, B. STAT3 Blockade with shRNA Enhances Radiosensitivity in Hep-2 Human Laryngeal Squamous Carcinoma Cells. Oncol. Rep. 2009, 23, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Vageli, D.P.; Doukas, P.G.; Siametis, A.; Judson, B.L. Targeting STAT3 Prevents Bile Reflux-induced Oncogenic Molecular Events Linked to Hypopharyngeal Carcinogenesis. J. Cell. Mol. Med. 2022, 26, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA Induces Fas-Mediated Apoptosis in Vitro and in Vivo in Breast Cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar]
- Pawlus, M.R.; Wang, L.; Hu, C.-J. STAT3 and HIF1α Cooperatively Activate HIF1 Target Genes in MDA-MB-231 and RCC4 Cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef]
- Ok Lee, S.; Lou, W.; Qureshi, K.M.; Mehraein-Ghomi, F.; Trump, D.L.; Gao, A.C. RNA Interference Targeting Stat3 Inhibits Growth and Induces Apoptosis of Human Prostate Cancer Cells. Prostate 2004, 60, 303–309. [Google Scholar] [CrossRef]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.-L.; Zhang, Z.; White, S.V.; Swiderski, P.; Lu, X.; DePinho, R.A.; Pal, S.K.; et al. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. 2018, 24, 5948–5962. [Google Scholar] [CrossRef]
- Huang, F.; Tong, X.; Fu, L.; Zhang, R. Knockdown of STAT3 by shRNA Inhibits the Growth of CAOV3 Ovarian Cancer Cell Line in Vitro and in Vivo. Acta Biochim. Biophys. Sin. 2008, 40, 519–525. [Google Scholar] [CrossRef]
- Wen, W.; Liang, W.; Wu, J.; Kowolik, C.M.; Buettner, R.; Scuto, A.; Hsieh, M.-Y.; Hong, H.; Brown, C.E.; Forman, S.J.; et al. Targeting JAK1/STAT3 Signaling Suppresses Tumor Progression and Metastasis in a Peritoneal Model of Human Ovarian Cancer. Mol. Cancer Ther. 2014, 13, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic Significance of STAT3 Expression and Its Correlation with Chemoresistance of Non-Small Cell Lung Cancer Cells. Acta Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.-J.; Jin, F.-G.; Liu, T.-G.; Fu, E.-Q.; Xie, Y.-H.; Sun, R.-L. Overexpression of STAT3 Potentiates Growth, Survival, and Radioresistance of Non-Small-Cell Lung Cancer (NSCLC) Cells. J. Surg. Res. 2011, 171, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Lu, X.; Di, B. Silencing STAT3 with Short Hairpin RNA Enhances Radiosensitivity of Human Laryngeal Squamous Cell Carcinoma Xenografts in Vivo. Exp. Ther. Med. 2010, 1, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Luwor, R.B.; Baradaran, B.; Taylor, L.E.; Iaria, J.; Nheu, T.V.; Amiry, N.; Hovens, C.M.; Wang, B.; Kaye, A.H.; Zhu, H.-J. Targeting Stat3 and Smad7 to Restore TGF-β Cytostatic Regulation of Tumor Cells in Vitro and in Vivo. Oncogene 2013, 32, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, L.; Li, Y.; Lin, G.; Shao, Y.; Ji, K.; Yu, H.; Hu, J.; Kalvakolanu, D.V.; Kopecko, D.J.; et al. Effects of Plasmid-Based Stat3-Specific Short Hairpin RNA and GRIM-19 on PC-3M Tumor Cell Growth. Clin. Cancer Res. 2008, 14, 559–568. [Google Scholar] [CrossRef]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 Activation in HER2-Overexpressing Breast Cancer Promotes Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, B.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. Targeting Blockage of STAT3 Inhibits Hepatitis B Virus-Related Hepatocellular Carcinoma. Cancer Biol. Ther. 2016, 17, 449–456. [Google Scholar] [CrossRef]
- Akiyama, Y.; Iizuka, A.; Kume, A.; Komiyama, M.; Urakami, K.; Ashizawa, T.; Miyata, H.; Omiya, M.; Kusuhara, M.; Yamaguchi, K. Effect of STAT3 Inhibition on the Metabolic Switch in a Highly STAT3-Activated Lymphoma Cell Line. Cancer Genom. Proteom. 2015, 12, 133. [Google Scholar]
- Li, W.-C.; Ye, S.-L.; Sun, R.-X.; Liu, Y.-K.; Tang, Z.-Y.; Kim, Y.; Karras, J.G.; Zhang, H. Inhibition of Growth and Metastasis of Human Hepatocellular Carcinoma by Antisense Oligonucleotide Targeting Signal Transducer and Activator of Transcription 3. Clin. Cancer Res. 2006, 12, 7140–7148. [Google Scholar] [CrossRef] [PubMed]
- Bielinska, A.; Shivdasani, R.A.; Zhang, L.; Nabel, G.J. Regulation of Gene Expression with Double-Stranded Phosphorothioate Oligonucleotides. Science 1990, 250, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Wei, H.; Tian, Z. STAT3-Decoy Oligodeoxynucleotide Inhibits the Growth of Human Lung Cancer via down-Regulating Its Target Genes. Oncol. Rep. 2007. [Google Scholar] [CrossRef]
- Njatcha, C.; Farooqui, M.; Kornberg, A.; Johnson, D.E.; Grandis, J.R.; Siegfried, J.M. STAT3 Cyclic Decoy Demonstrates Robust Antitumor Effects in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2018, 17, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, P.; Zhang, B.; Wang, A.; Yang, M. Role of STAT3 Decoy Oligodeoxynucleotides on Cell Invasion and Chemosensitivity in Human Epithelial Ovarian Cancer Cells. Cancer Genet. Cytogenet. 2010, 197, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, F.; Wen, Z.; Shi, M.; Zhang, H. Blockage of STAT3 Signaling Pathway with a Decoy Oligodeoxynucleotide Inhibits Growth of Human Ovarian Cancer Cells. Cancer Investig. 2014, 32, 8–12. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhang, B.; Mao, H.; Shen, L.; Ma, Y. Inhibitory Effects of STAT3 Decoy Oligodeoxynucleotides on Human Epithelial Ovarian Cancer Cell Growth in Vivo. Int. J. Mol. Med. 2013, 32, 623–628. [Google Scholar] [CrossRef]
- Gu, J.; Li, G.; Sun, T.; Su, Y.; Zhang, X.; Shen, J.; Tian, Z.; Zhang, J. Blockage of the STAT3 Signaling Pathway with a Decoy Oligonucleotide Suppresses Growth of Human Malignant Glioma Cells. J. Neurooncol. 2008, 89, 9–17. [Google Scholar] [CrossRef]
- Shen, J.; Li, R.; Li, G. Inhibitory Effects of Decoy-ODN Targeting Activated STAT3 on Human Glioma Growth in Vivo. In Vivo 2009, 23, 237–243. [Google Scholar]
- Sun, X.; Zhang, J.; Wang, L.; Tian, Z. Growth Inhibition of Human Hepatocellular Carcinoma Cells by Blocking STAT3 Activation with Decoy-ODN. Cancer Lett. 2008, 262, 201–213. [Google Scholar] [CrossRef]
- Asadi, Z.; Fathi, M.; Rismani, E.; Bigdelou, Z.; Johari, B. Application of Decoy Oligodeoxynucleotides Strategy for Inhibition of Cell Growth and Reduction of Metastatic Properties in Nonresistant and Erlotinib-resistant SW480 Cell Line. Cell Biol. Int. 2021, 45, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-Human Trial of a STAT3 Decoy Oligonucleotide in Head and Neck Tumors: Implications for Cancer Therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-W.; Yang, S.-T.; Chien, M.-H.; Hua, K.-T.; Wu, C.-J.; Hsiao, S.M.; Lin, H.; Hsiao, M.; Su, J.-L.; Wei, L.-H. The STAT3-miRNA-92-Wnt Signaling Pathway Regulates Spheroid Formation and Malignant Progression in Ovarian Cancer. Cancer Res. 2017, 77, 1955–1967. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, C.; Zhou, W.; Wu, C.; Zhang, Y. The STAT3/NFIL3 Signaling Axis-mediated Chemotherapy Resistance Is Reversed by Raddeanin A via Inducing Apoptosis in Choriocarcinoma Cells. J. Cell. Physiol. 2018, 233, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-Y.; Lin, H.-C.; Tsai, F.-C.; Ko, J.-Y.; Kok, S.-H.; Cheng, S.-J.; Lee, J.-J.; Chia, J.-S. Effects of Interleukin-6 on STAT3-Regulated Signaling in Oral Cancer and as a Prognosticator of Patient Survival. Oral. Oncol. 2022, 124, 105665. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Arlinghaus, R.B. Knockdown of STAT3 Expression by RNA Interference Inhibits the Induction of Breast Tumors in Immunocompetent Mice. Cancer Res. 2005, 65, 2532–2536. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Huang, C.; Cao, J.; Huang, K.-J.; Jiang, T.; Qiu, Z.-J. Lentivirus-Mediated shRNA Interference Targeting STAT3 Inhibits Human Pancreatic Cancer Cell Invasion. WJG 2009, 15, 3757. [Google Scholar] [CrossRef]
- Li, H.D.; Huang, C.; Huang, K.J.; Wu, W.D.; Jiang, T.; Cao, J.; Feng, Z.Z.; Qiu, Z.J. STAT3 Knockdown Reduces Pancreatic Cancer Cell Invasiveness and Matrix Metalloproteinase-7 Expression in Nude Mice. PLoS ONE 2011, 6, e25941. [Google Scholar] [CrossRef]
- Qian, W.F.; Guan, W.X.; Gao, Y.; Tan, J.F.; Qiao, Z.M.; Huang, H.; Xia, C.L. Inhibition of STAT3 by RNA Interference Suppresses Angiogenesis in Colorectal Carcinoma. Braz. J. Med. Biol. Res. 2011, 44, 1222–1230. [Google Scholar] [CrossRef]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.-J.; Li, P.-K.; Li, C.; Lin, J. STAT3 Is Necessary for Proliferation and Survival in Colon Cancer–Initiating Cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef]
- Xu, D.; Chen, S.; Zhou, P.; Wang, Y.; Zhao, Z.; Wang, X.; Huang, H.; Xue, X.; Liu, Q.; Wang, Y.; et al. Suppression of Esophageal Cancer Stem-like Cells by SNX-2112 Is Enhanced by STAT3 Silencing. Front. Pharmacol. 2020, 11, 532395. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Das, S.; Paul, A.; Samadder, A.; Bhattacharyya, S.S.; Khuda-Bukhsh, A.R. Assessment of Drug Delivery and Anticancer Potentials of Nanoparticles-Loaded siRNA Targeting STAT3 in Lung Cancer, in Vitro and in Vivo. Toxicol. Lett. 2014, 225, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, C.; Zhang, L.; Chen, Y.; Ye, Y.; Zhao, M. Knockdown of STAT3 Expression in SKOV3 Cells by Biodegradable siRNA–PLGA/CSO Conjugate Micelles. Colloids Surf. B Biointerfaces 2015, 127, 155–163. [Google Scholar] [CrossRef]
- Zhang, H.; Men, K.; Pan, C.; Gao, Y.; Li, J.; Lei, S.; Zhu, G.; Li, R.; Wei, Y.; Duan, X. Treatment of Colon Cancer by Degradable rrPPC Nano-Conjugates Delivered STAT3 siRNA. Int. J. Nanomed. 2020, 15, 9875–9890. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Mansourian, M.; Koning, G.A.; Badiee, A.; Jaafari, M.R.; Ten Hagen, T.L.M. Development of a Novel Cyclic RGD Peptide for Multiple Targeting Approaches of Liposomes to Tumor Region. J. Control. Release 2015, 220, 308–315. [Google Scholar] [CrossRef]
- Joshi, N.; Hajizadeh, F.; Ansari Dezfouli, E.; Zekiy, A.O.; Nabi Afjadi, M.; Mousavi, S.M.; Hojjat-Farsangi, M.; Karpisheh, V.; Mahmoodpoor, A.; Hassannia, H.; et al. Silencing STAT3 Enhances Sensitivity of Cancer Cells to Doxorubicin and Inhibits Tumor Progression. Life Sci. 2021, 275, 119369. [Google Scholar] [CrossRef]
- Malhotra, M.; Tomaro-Duchesneau, C.; Prakash, S. Synthesis of TAT Peptide-Tagged PEGylated Chitosan Nanoparticles for siRNA Delivery Targeting Neurodegenerative Diseases. Biomaterials 2013, 34, 1270–1280. [Google Scholar] [CrossRef]
- Dai, L.; Cheng, L.; Zhang, X.; Jiang, Q.; Zhang, S.; Wang, S.; Li, Y.; Chen, X.; Du, T.; Yang, Y.; et al. Plasmid-Based STAT3-siRNA Efficiently Inhibits Breast Tumor Growth and Metastasis in Mice. Neoplasma 2011, 58, 538–547. [Google Scholar] [CrossRef]
- Tian, W.; Li, B.; Zhang, X.; Dang, W.; Wang, X.; Tang, H.; Wang, L.; Cao, H.; Chen, T. Suppression of Tumor Invasion and Migration in Breast Cancer Cells Following Delivery of siRNA against Stat3 with the Antimicrobial Peptide PR39. Oncol. Rep. 2012, 28, 1362–1368. [Google Scholar] [CrossRef]
- Luo, K.; Gao, Y.; Yin, S.; Yao, Y.; Yu, H.; Wang, G.; Li, J. Co-Delivery of Paclitaxel and STAT3 siRNA by a Multifunctional Nanocomplex for Targeted Treatment of Metastatic Breast Cancer. Acta Biomater. 2021, 134, 649–663. [Google Scholar] [CrossRef]
- Shakeran, Z.; Varshosaz, J.; Keyhanfar, M.; Mohammad-Beigi, H.; Rahimi, K.; Sutherland, D.S. Co-Delivery of STAT3 siRNA and Methotrexate in Breast Cancer Cells. Artif. Cells Nanomed. Biotechnol. 2022, 50, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Abdelmoaty, M.M.; Curran, S.M.; Dyavar, S.R.; Kumar, D.; Alnouti, Y.; Coulter, D.W.; Podany, A.T.; Singh, R.K.; Vetro, J.A. Direct Comparison of Chol-siRNA Polyplexes and Chol-DsiRNA Polyplexes Targeting STAT3 in a Syngeneic Murine Model of TNBC. ncRNA 2022, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.; Labala, S.; Ninave, K.M.; Gade, S.K.; Venuganti, V.V.K. Effective Skin Cancer Treatment by Topical Co-Delivery of Curcumin and STAT3 siRNA Using Cationic Liposomes. AAPS PharmSciTech 2018, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Erdene-Ochir, T.; Ganbold, T.; Zandan, J.; Han, S.; Borjihan, G.; Baigude, H. Alkylation Enhances Biocompatibility and siRNA Delivery Efficiency of Cationic Curdlan Nanoparticles. Int. J. Biol. Macromol. 2020, 143, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ruan, W.; Qin, M.; Long, Y.; Wan, T.; Yu, K.; Zhai, Y.; Wu, C.; Xu, Y. Intradermal Delivery of STAT3 siRNA to Treat Melanoma via Dissolving Microneedles. Sci. Rep. 2018, 8, 1117. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Dai, L.; Cheng, L.; Chen, X.; Li, Y.; Zhang, S.; Su, X.; Zhao, X.; Wei, Y.; Deng, H. Efficient Inhibition of Intraperitoneal Ovarian Cancer Growth in Nude Mice by Liposomal Delivery of Short Hairpin RNA against STAT 3. J. Obs. Gynaecol. Res. 2013, 39, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Jiang, Z.; Sun, L.; Wang, L.; Gu, Z.; Li, W.; Guo, L.; Chen, X.; Zhang, H.; et al. Effects on the STAT3-shRNA in Non-Small-Cell Lung Cancer Therapy: Design, Induction of Apoptosis, and Conjugation with Chitosan-Based Gene Vectors. J. Ocean. Univ. China 2021, 20, 1097–1108. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Fan, X.; Mao, D.; Lv, S.; Chen, P. Effect of STAT3 Decoy Oligodeoxynucleotides Mediated by Ultrasound-Targeted Microbubbles Combined with Ultrasound on the Growth of Squamous Cell Carcinoma of the Esophagus. Oncol. Lett. 2018, 17, 2151–2158. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, T.; Ma, Y.; Li, R.; Pang, Y.; Mao, H.; Liu, P. Novel Nanocomplexes Targeting STAT3 Demonstrate Promising Anti-Ovarian Cancer Effects in Vivo. OncoTargets Ther. 2020, 13, 5069–5082. [Google Scholar] [CrossRef]
- Shi, K.; Fang, Y.; Gao, S.; Yang, D.; Bi, H.; Xue, J.; Lu, A.; Li, Y.; Ke, L.; Lin, X.; et al. Inorganic Kernel—Supported Asymmetric Hybrid Vesicles for Targeting Delivery of STAT3-Decoy Oligonucleotides to Overcome Anti-HER2 Therapeutic Resistance of BT474R. J. Control. Release 2018, 279, 53–68. [Google Scholar] [CrossRef]
- City of Hope Medical Center A Phase I Study of Intratumoral Injections of CpG-STAT3 siRNA (CAS3/SS3) in Combination with Local Radiation in Patients With Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma (NHL). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04995536?term=NCT04995536&draw=2&rank=1 (accessed on 28 December 2022).
- Home—ClinicalTrials.Gov. Available online: https://classic.clinicaltrials.gov/ct2/home (accessed on 28 December 2022).
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 Antisense Oligonucleotide AZD9150 in a Subset of Patients with Heavily Pretreated Lymphoma: Results of a Phase 1b Trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Acerta Pharma BV PRISM: A Platform Protocol for the Treatment of Relapsed/Refractory Aggressive Non-Hodgkin’s Lymphoma. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03527147?term=NCT03527147&draw=2&rank=1 (accessed on 28 December 2022).
- AstraZeneca A Phase I, Open-Label Study to Assess the Safety, Tolerability, Pharmacokinetics and Anti-Tumor Activity of AZD9150 Monotherapy and AZD9150 in Combination with Durvalumab in Japanese Patients with Advanced Solid Malignancies. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03394144?term=NCT03394144&draw=2&rank=1 (accessed on 28 December 2022).
- Nishina, T.; Fujita, T.; Yoshizuka, N.; Sugibayashi, K.; Murayama, K.; Kuboki, Y. Safety, Tolerability, Pharmacokinetics and Preliminary Antitumour Activity of an Antisense Oligonucleotide Targeting STAT3 (Danvatirsen) as Monotherapy and in Combination with Durvalumab in Japanese Patients with Advanced Solid Malignancies: A Phase 1 Study. BMJ Open 2022, 12, e055718. [Google Scholar] [CrossRef] [PubMed]
- MedImmune LLC A Phase 1b Study to Evaluate the Safety and Efficacy of MEDI4736 as Monotherapy and in Combination with Tremelimumab or AZD9150 in Subjects with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02549651?term=NCT02549651&draw=2&rank=1 (accessed on 28 December 2022).
- Cascone, T.; García-Campelo, R.; Spicer, J.; Weder, W.; Daniel, D.; Spigel, D.; Hussein, M.; Mazieres, J.; Oliveira, J.; Yau, E.; et al. Abstract CT011: NeoCOAST: Open-Label, Randomized, Phase 2, Multidrug Platform Study of Neoadjuvant Durvalumab Alone or Combined with Novel Agents in Patients (Pts) with Resectable, Early-Stage Non-Small-Cell Lung Cancer (NSCLC). Cancer Res. 2022, 82, CT011. [Google Scholar] [CrossRef]
- Campelo, R.G.; Forde, P.; Weder, W.; Spicer, J.; He, P.; Hamid, O.; Martinez, P.; Cascone, T. P2.04-28 NeoCOAST: Neoadjuvant Durvalumab Alone or with Novel Agents for Resectable, Early-Stage (I–IIIA) Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, S719. [Google Scholar] [CrossRef]
- AstraZeneca A Phase 1b/2, Open-Label, Multicentre Study Assessing the Safety, Tolerability, Pharmacokinetics, and Preliminary Anti-Tumor Activity of MEDI4736 in Combination with AZD9150 or AZD5069 in Patients with Advanced Solid Malignancies and Subsequently Comparing AZD9150 and AZD5069 Both as Monotherapy and in Combination with MEDI4736 as Second Line Treatment in Patients with Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02499328?term=NCT02499328&draw=2&rank=1 (accessed on 28 December 2022).
- AstraZeneca A Phase Ib/II, Open-Label, Multicentre Study to Assess Safety, Tolerability, Pharmacokinetics and Preliminary Anti-Tumour Activity of AZD9150 Plus Durvalumab Alone or in Combination with Chemotherapy in Patients with Advanced, Solid Tumours and Subsequently in Patients with Non-Small-Cell Lung Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03421353?term=NCT03421353&draw=2&rank=1 (accessed on 28 December 2022).
- Phase II Umbrella Study of Novel Anti-Cancer Agents in Patients with NSCLC Who Progressed on an Anti-PD-1/PD-L1 Containing Therapy—Full Text View. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03334617?term=NCT03334617&draw=2&rank=1 (accessed on 28 December 2022).
- A Pilot Study of AZD9150, a STAT3 Antisense Oligonucleotide in Malignant Ascites. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02417753?term=NCT02417753&draw=1&rank=1 (accessed on 28 December 2022).
- University of Pittsburgh Preliminary Assessment of the Safety and Biological Activity of Intratumoral STAT3 DECOY in Surgically Resectable Head and Neck Squamous Cell Carcinoma. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT00696176?term=NCT00696176&draw=2&rank=1 (accessed on 28 December 2022).
- Lau, K.Y.-T.; Ramaiyer, M.; Johnson, D.E.; Grandis, R.J. Targeting STAT3 in Cancer with Nucleotide Therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Abdel-Aziz, M. STAT3 Transcription Factor as Target for Anti-Cancer Therapy. Pharmacol. Rep. 2020, 72, 1101–1124. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Guan, X.; Qin, J.-J. Inhibiting STAT3 Signaling Pathway by Natural Products for Cancer Prevention and Therapy: In Vitro and in Vivo Activity and Mechanisms of Action. Pharmacol. Res. 2022, 182, 106357. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Targeting STAT3 Signaling Pathway in Colorectal Cancer. Biomedicines 2021, 9, 1016. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Florczak, A.; Deptuch, T.; Lewandowska, A.; Penderecka, K.; Kramer, E.; Marszalek, A.; Mackiewicz, A.; Dams-Kozlowska, H. Functionalized Silk Spheres Selectively and Effectively Deliver a Cytotoxic Drug to Targeted Cancer Cells in Vivo. J. Nanobiotechnol. 2020, 18, 177. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Florczak, A.; Smialek, M.; Dondajewska, E.; Mackiewicz, A.; Kortylewski, M.; Dams-Kozlowska, H. Functionalized Bioengineered Spider Silk Spheres Improve Nuclease Resistance and Activity of Oligonucleotide Therapeutics Providing a Strategy for Cancer Treatment. Acta Biomater. 2017, 59, 221–233. [Google Scholar] [CrossRef] [PubMed]



| Oligo | Cancer | In Vitro Study | In Vivo Study | Ref. | ||
|---|---|---|---|---|---|---|
| Cell Line /Type of Viral Vector | Effect | Cell Line /Route of Viral Vector Administration | Effect | |||
| shRNA | Ovarian cancer | SKOV3 /Lentiviral vector | Spheroid formation ↓ | SKOV3 /in vitro modified cells | Metastasis ↓ Tumor growth ↓ | [177] |
| Choriocarcinoma | JEG-3 /Lentiviral vector | Chemoresistance ↓ | nd | nd | [178] | |
| Oral cancer | SAS /Lentiviral vector | Proliferation ↓ | SAS /in vitro modified cells | Tumor growth ↓ | [179] | |
| Breast cancer | 4T1 /Lentiviral vector | Invasiveness ↓ | 4T1 /in vitro modified cells | Metastasis ↓ Tumor growth ↓ | [180] | |
| Pancreatic cancer | SW1990 /Lentiviral vector | Proliferation ↓ Invasion potential ↓ | SW1990 /in vitro modified cells | Tumor growth ↓ Angiogenesis ↓ | [181,182] | |
| Colorectal carcinoma | HT-29 /Lentiviral vector | Proliferation ↓ | HT-29 /in vitro modified cells | Tumor growth ↓ Angiogenesis ↓ | [183] | |
| Colon cancer | SW480 HCT116 /Lentiviral vector | Viability ↓ | HCT116 /in vitro modified cells | Tumor growth ↓ | [184] | |
| Esophageal cancer | Eca109 HEEC /Lentiviral vector | Proliferation ↓ Colony formation ↓ Chemoresistance ↓ Cell cycle arrest | Eca109 HEEC /in vitro modified cells | Apoptosis ↑ Tumor growth ↓ Chemoresistance ↓ | [185] | |
| Oligo | Status | Phase | Start Date | Completion Date | Disease | Enrollment | Study Identifier | Sponsor/Collaborator | Location Countries | Combination |
|---|---|---|---|---|---|---|---|---|---|---|
| ASO AZD9150 | Completed | I/II | 27 February 2012 | 23 March 2016 | DLBCL Lymphoma Advanced cancers | 64 | NCT01563302 | Ionis Pharmaceuticals Inc. AstraZeneca | USA | None |
| Completed | I/Ib | May 2013 | February 2015 | Advanced and metastatic HCC | 58 | NCT01839604 | AstraZeneca Ionis Pharmaceuticals Inc. | Hong Kong, Japan, South Korea, Taiwan | None | |
| Terminated (due to inability to find eligible patients) | II | 3 April 2015 | 7 April 2016 | Ovarian cancer Gastrointestinal cancer ascites | 1 | NCT02417753 | National Cancer Institute (NCI) | USA | None | |
| Active, not recruiting | Ib/II | 6 August 2015 | 29 December 2023 | HNSCC Advanced solid tumors | 340 | NCT02499328 | AstraZeneca MedImmune LLC | USA, Belgium, Germany, Italy, Spain, UK | Durvalumab | |
| Completed | Ib | 13 July 2016 | 4 February 2019 | DLBCL | 32 | NCT02549651 | MedImmune LLC | USA, France, Ireland, UK | Durvalumab | |
| Active, not recruiting | I | 3 October 2016 | 31 March 2023 | Muscle invasive Bladder cancer | 156 | NCT02546661 | AstraZeneca | USA, Canada, France, Spain, UK | Durvalumab | |
| Active, not recruiting | II | 2 March 2017 | 31 March 2023 | Pancreatic cancer Colorectal cancer NSCLC | 53 | NCT02983578 | M.D. Anderson Cancer Center National Cancer Institute (NCI) AstraZeneca | USA | Durvalumab | |
| Recruiting | II | 18 December 2017 | 2 January 2026 | Metastatic NSCLC | 530 | NCT03334617 | AstraZeneca | USA, Austria, Canada, France, Germany, Israel, South Korea, Spain | Durvalumab | |
| Completed | I | 30 January 2018 | 12 April 2019 | Advanced solid malignancies | 11 | NCT03394144 | AstraZeneca | Japan | Durvalumab | |
| Active, not recruiting | Ib/II | 7 February 2018 | 31 December 2025 | NSCLC Advanced solid tumors | 76 | NCT03421353 | AstraZeneca | USA | Durvalumab Chemotherapy: Cisplatin/ 5-Flourouracil/ Carboplatin/ Gemcitabine/ Nab-paclitaxel | |
| Completed | Ib | 19 June 2018 | 31 March 2021 | NHL DLBCL | 30 | NCT03527147 | Acerta Pharma BV AstraZeneca | USA | Acalabrutinib | |
| Active, not recruiting | Ib | 27 December 2018 | 26 March 2026 | Metastatic NSCLC | 258 | NCT03819465 | AstraZeneca | USA, Austria, Belgium, Canada, Korea, Poland Spain, Russia, Taiwan, Thailand | Durvalumab Chemotherapy: Pemetrexed/ Carboplatin/ Gemcitabine/ Cisplatin/ Nab-paclitaxel | |
| Completed | II | 8 March 2019 | 13 January 2021 | Early-stage NSCLC | 84 | NCT03794544 | MedImmune LLC | USA, Canada, France, Italy, Portugal, Spain, Switzerland | Durvalumab | |
| ODN-decoy | Completed | 0 | August 2008 | August 2011 | Head and neck cancer | 32 | NCT00696176 | University of Pittsburgh | USA | None |
| CpG-siRNA | Recruiting | I | 1 August 2022 | 27 January 2024 | Lymphoma | 18 | NCT04995536 | City of Hope Medical Center National Cancer Institute (NCI) | USA | Radiation therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molenda, S.; Sikorska, A.; Florczak, A.; Lorenc, P.; Dams-Kozlowska, H. Oligonucleotide-Based Therapeutics for STAT3 Targeting in Cancer—Drug Carriers Matter. Cancers 2023, 15, 5647. https://doi.org/10.3390/cancers15235647
Molenda S, Sikorska A, Florczak A, Lorenc P, Dams-Kozlowska H. Oligonucleotide-Based Therapeutics for STAT3 Targeting in Cancer—Drug Carriers Matter. Cancers. 2023; 15(23):5647. https://doi.org/10.3390/cancers15235647
Chicago/Turabian StyleMolenda, Sara, Agata Sikorska, Anna Florczak, Patryk Lorenc, and Hanna Dams-Kozlowska. 2023. "Oligonucleotide-Based Therapeutics for STAT3 Targeting in Cancer—Drug Carriers Matter" Cancers 15, no. 23: 5647. https://doi.org/10.3390/cancers15235647
APA StyleMolenda, S., Sikorska, A., Florczak, A., Lorenc, P., & Dams-Kozlowska, H. (2023). Oligonucleotide-Based Therapeutics for STAT3 Targeting in Cancer—Drug Carriers Matter. Cancers, 15(23), 5647. https://doi.org/10.3390/cancers15235647