
Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
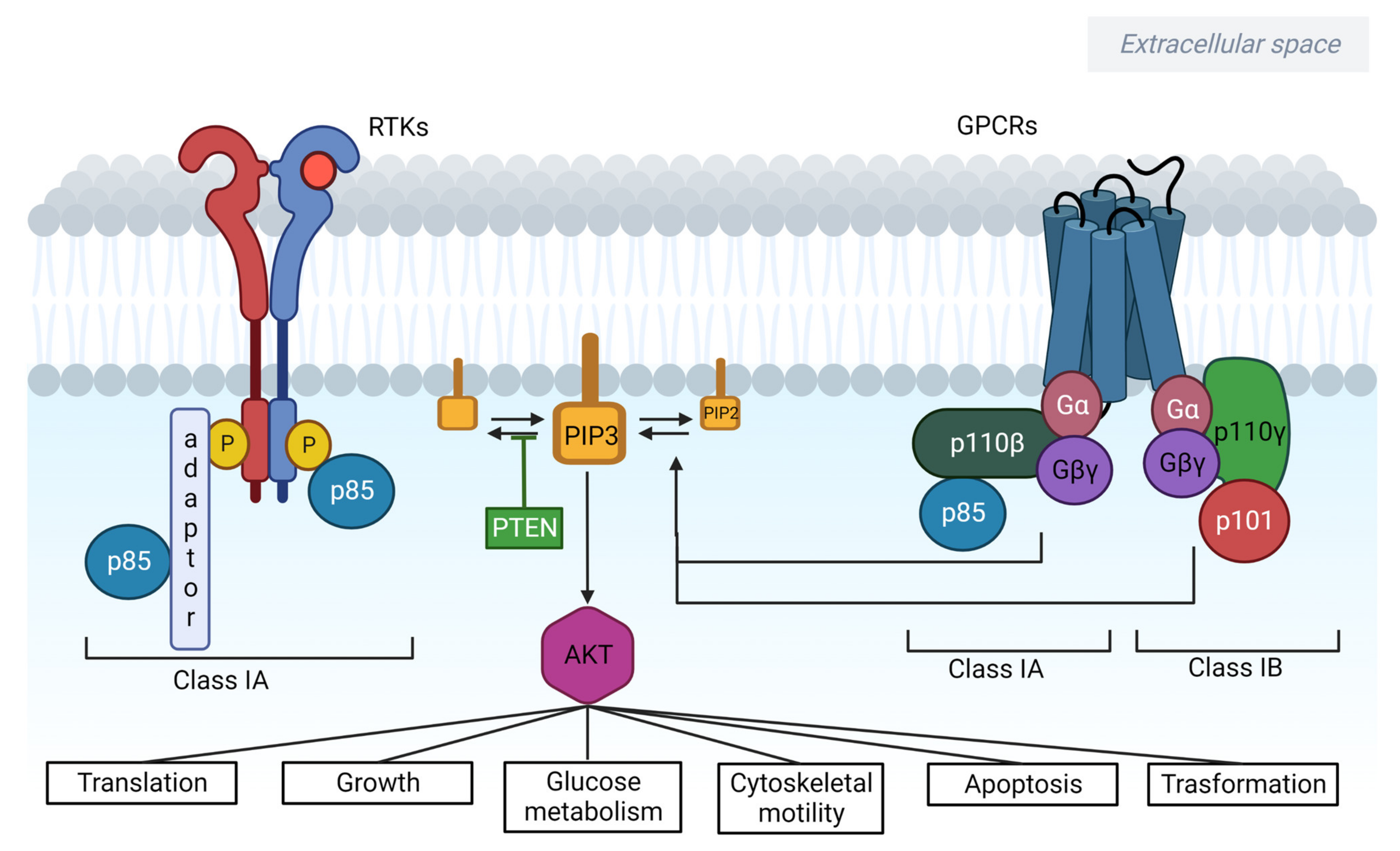
2. PI3K/AKT/mTOR Signalling in Cancer
2.1. PI3Ks
2.1.1. Class I
2.1.2. Class II
2.1.3. Class III
2.2. AKT
2.3. PTEN
2.4. mTOR
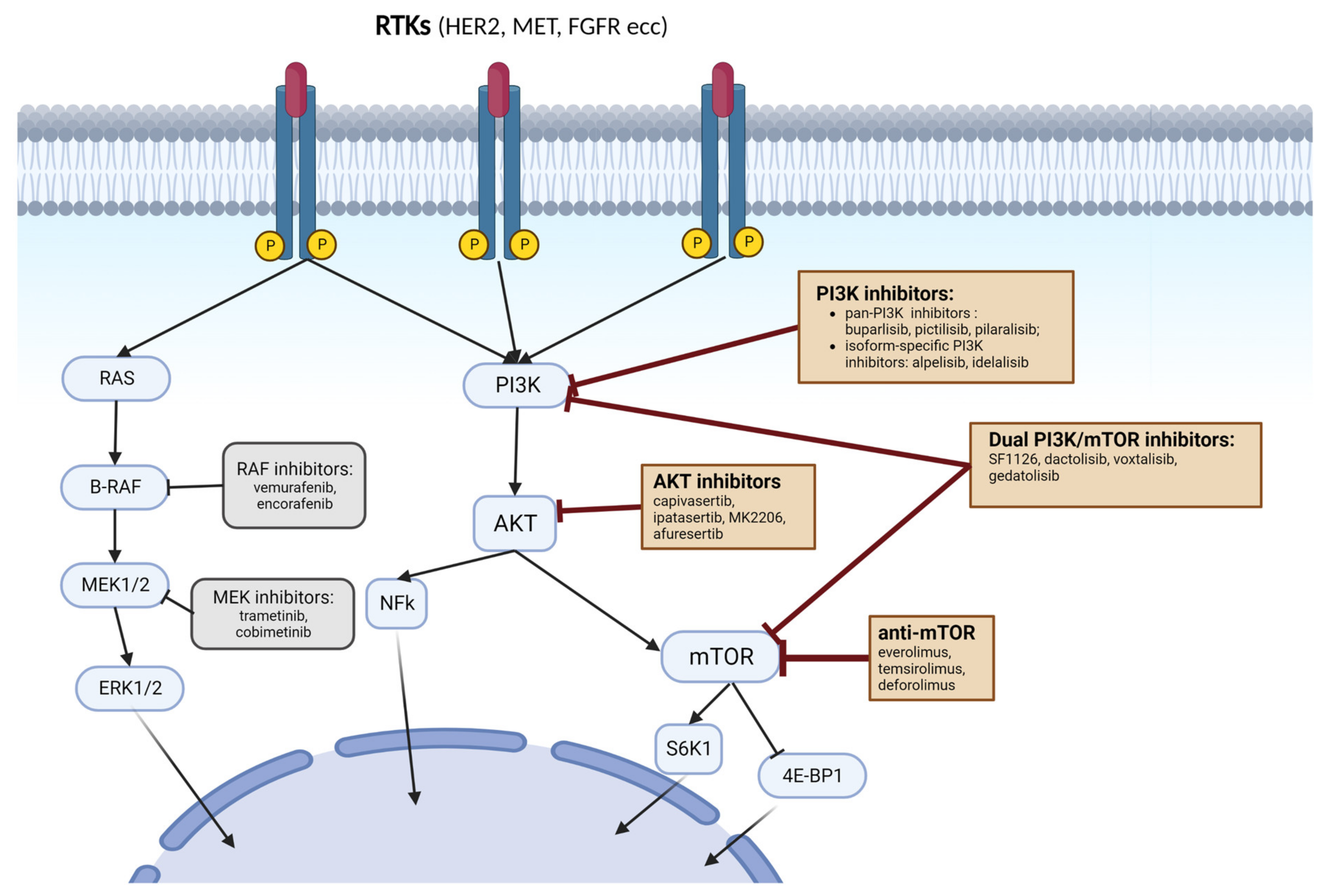
3. Targeting PI3K Pathway in Cancer
3.1. PI3K Inhibitors
3.1.1. Pan-PI3K Inhibitors
3.1.2. Isoform-Selective PI3K Inhibitors
3.2. AKT Inhibitors
3.3. mTORC1 and mTORC2 Inhibitors
3.3.1. ATP-Competitive mTOR Inhibitors
3.3.2. Dual PI3K/mTOR Inhibitors
3.4. Combination Strategies
3.4.1. Her-2 Inhibitors
3.4.2. MAPK Inhibitors
3.4.3. Chemotherapy
3.4.4. Immunotherapy
4. Impact of Biomarkers
5. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A. Targeting PI3K Signalling in Cancer: Opportunities, Challenges and Limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.; Mandala, M. PI3K-AKT-mTOR Inhibition in Cancer Immunotherapy, Redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, A.; Vera-Badillo, F.; Al-Mubarak, M.; Templeton, A.J.; Corrales-Sánchez, V.; Díez-González, L.; Cuenca-Lopez, M.D.; Seruga, B.; Pandiella, A.; Amir, E. Activation of the PI3K/mTOR/AKT Pathway and Survival in Solid Tumors: Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e95219. [Google Scholar] [CrossRef]
- Du Rusquec, P.; Blonz, C.; Frenel, J.S.; Campone, M. Targeting the PI3K/Akt/mTOR Pathway in Estrogen-Receptor Positive HER2 Negative Advanced Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920940939. [Google Scholar] [CrossRef]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR Signaling Pathway as a Therapeutic Target for Ovarian Cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef]
- Dobashi, Y.; Watanabe, Y.; Miwa, C.; Suzuki, S.; Koyama, S. Mammalian Target of Rapamycin: A Central Node of Complex Signaling Cascades. Int. J. Clin. Exp. Pathol. 2011, 4, 476–495. [Google Scholar]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K Pathway in Cancer: Are We Making Headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Saal, L.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent Gross Mutations of the PTEN Tumor Suppressor Gene in Breast Cancers with Deficient DSB Repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.K.; Lopez-Knowles, E.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol Polyphosphate 4-Phosphatase II Regulates PI3K/Akt Signaling and is Lost in Human Basal-Like Breast Cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.W.; Mills, G.B. Targeting Therapeutic Liabilities Engendered by PIK3R1 Mutations for Cancer Treatment. Pharmacogenomics 2016, 17, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Jean, S.; Kiger, A.A. Classes of Phosphoinositide 3-Kinases at a Glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Toker, A.; Cantley, L.C. Signalling through the Lipid Products of Phosphoinositide-3-OH Kinase. Nature 1997, 387, 673–676. [Google Scholar] [CrossRef]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-Yl)-6-(4-Methanesulfonyl-Piperazin-1-Ylmethyl)-4-Morpholin-4-Yl-Thieno[3,2-d]Pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef]
- Burke, J.E. Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Kriplani, N.; Hermida, M.A.; Brown, E.R.; Leslie, N.R. Class I PI 3-Kinases: Function and Evolution. Adv. Biol. Regul. 2015, 59, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; PCAWG Evolution & Heterogeneity Working Group; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; et al. The Evolutionary History of 2658 Cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B.B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-Kinase-δ Inhibitor CAL-101 Shows Promising Preclinical Activity in Chronic Lymphocytic Leukemia by Antagonizing Intrinsic and Extrinsic Cellular Survival Signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Tsolakos, N.; Durrant, T.N.; Chessa, T.; Suire, S.M.; Oxley, D.; Kulkarni, S.; Downward, J.; Perisic, O.; Williams, R.L.; Stephens, L.; et al. Quantitation of Class IA PI3Ks in Mice Reveals P110-Free-P85s and Isoform-Selective Subunit Associations and Recruitment to Receptors. Proc. Natl. Acad. Sci. USA 2018, 115, 12176–12181. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ Is a Rab5 Effector Selectively Controlling Endosomal Akt2 Activation Downstream of Insulin Signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [Green Version]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2α Scaffolding Function. Cancer Cell 2017, 32, 444–459.e7. [Google Scholar] [CrossRef] [Green Version]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef]
- Marat, A.L.; Haucke, V. Phosphatidylinositol 3-Phosphates—At the Interface between Cell Signalling and Membrane Traffic. EMBO J. 2016, 35, 561–579. [Google Scholar] [CrossRef]
- O’Farrell, F.; Lobert, V.H.; Sneeggen, M.; Jain, A.; Katheder, N.; Wenzel, E.M.; Schultz, S.W.; Tan, K.W.; Brech, A.; Stenmark, H.; et al. Class III Phosphatidylinositol-3-OH Kinase Controls Epithelial Integrity through Endosomal LKB1 Regulation. Nat. Cell Biol. 2017, 19, 1412–1423. [Google Scholar] [CrossRef] [Green Version]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 Kinase Domain Dynamics Regulate the Autophagic PI 3-Kinase Complex. Mol. Cell 2017, 67, 528–534.e3. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular Cloning of the Akt Oncogene and Its Human Homologues AKT1 and AKT2: Amplification of AKT1 in a Primary Human Gastric Adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [Green Version]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT Pathway in Cancer: The Framework of Malignant Behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [Green Version]
- Cisse, O.; Quraishi, M.; Gulluni, F.; Guffanti, F.; Mavrommati, I.; Suthanthirakumaran, M.; Oh, L.C.R.; Schlatter, J.N.; Sarvananthan, A.; Broggini, M.; et al. Downregulation of Class II Phosphoinositide 3-Kinase PI3K-C2β Delays Cell Division and Potentiates the Effect of Docetaxel on Cancer Cell Growth. J. Exp. Clin. Cancer Res. 2019, 38, 472. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting mTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene 2021, 40, 5590–5599. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, X.; Pintilie, M.; Ma, N.; Miller, N.; Banerjee, D.; Tsao, M.-S.; Mak, T.; Fyles, A.; Liu, F.-F. Dysregulated PTEN-PKB and Negative Receptor Status in Human Breast Cancer. Int. J. Cancer 2003, 104, 195–203. [Google Scholar] [CrossRef]
- Nunnery, S.; Mayer, I. Management of Toxicity to Isoform α-Specific PI3K Inhibitors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, x21–x26. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V. Development of PI3K/AKT/mTOR Pathway Inhibitors and Their Application in Personalized Therapy for Non-Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2012, 7, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, I.U.; Hodgson, M.C.; Bowden, W.A.; Ittmann, M.M. INPP4B: The New Kid on the PI3K Block. Oncotarget 2011, 2, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Naizabekov, S.; Chen, Z.; Tokay, T. Power of PTEN/AKT: Molecular Switch between Tumor Suppressors and Oncogenes. Oncol. Lett. 2016, 12, 375–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngeow, J.; Eng, C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb. Perspect. Med. 2019, 10, a036087. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Murugan, A.K. mTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Krencz, I.; Sebestyen, A.; Khoor, A. mTOR in Lung Neoplasms. Pathol. Oncol. Res. 2020, 26, 35–48. [Google Scholar] [CrossRef]
- Niederst, M.J.; Engelman, J.A. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Minjgee, M.; Saki, M.; Holler, M.; Meier, F.; Eicheler, W.; Rodemann, H.P. ERK2-Dependent Reactivation of Akt Mediates the Limited Response of Tumor Cells with Constitutive K-RAS Activity to PI3K Inhibition. Cancer Biol. Ther. 2014, 15, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Fekete, M.; Santiskulvong, C.; Eng, C.; Dorigo, O. Effect of PI3K/Akt Pathway Inhibition-Mediated G1 Arrest on Chemosensitization in Ovarian Cancer Cells. Anticancer. Res. 2012, 32, 445–452. [Google Scholar]
- Carden, C.P.; Stewart, A.; Thavasu, P.; Kipps, E.; Pope, L.; Crespo, M.; Miranda, S.; Attard, G.; Garrett, M.D.; Clarke, P.A.; et al. The Association of PI3 Kinase Signaling and Chemoresistance in Advanced Ovarian Cancer. Mol. Cancer Ther. 2012, 11, 1609–1617. [Google Scholar] [CrossRef]
- Garces, A.E.; Stocks, M.J. Class 1 PI3K Clinical Candidates and Recent Inhibitor Design Strategies: A Medicinal Chemistry Perspective. J. Med. Chem. 2019, 62, 4815–4850. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors as Cancer Therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-Independent HER2/HER3/PI3K Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the PI3K Inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan-Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Mayer, I.A.; Abramson, V.G.; Isakoff, S.J.; Forero, A.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Yap, J.; Van den Abbeele, A.D.; Li, Y.; et al. Stand up to Cancer Phase Ib Study of Pan-Phosphoinositide-3-Kinase Inhibitor Buparlisib with Letrozole in Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1202–1209. [Google Scholar] [CrossRef]
- Speranza, M.C.; Nowicki, M.O.; Behera, P.; Cho, C.-F.; Chiocca, E.A.; Lawler, S.E. BKM-120 (Buparlisib): A Phosphatidyl-Inositol-3 Kinase Inhibitor with Anti-Invasive Properties in Glioblastoma. Sci. Rep. 2016, 6, 20189. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; De Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 Study of Buparlisib (BKM120), a Pan-Class I PI3K Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of Phosphatidylinositol-3 Kinase Promotes Escape from Hormone Dependence in Estrogen Receptor—Positive Human Breast Cancer. J. Clin. Investig. 2010, 120, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the Phosphatidylinositol 3-Kinase Pathway: Role in Tumor Progression and Therapeutic Implications in Breast Cancer. Breast Cancer Res. BCR 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.-A.; Kalev, D.; Egle, D.; et al. Buparlisib plus Fulvestrant in Postmenopausal Women with Hormone-Receptor-Positive, HER2-Negative, Advanced Breast Cancer Progressing on or After mTOR Inhibition (BELLE-3): A randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Chan, A.; Dirix, L.; O’Shaughnessy, J.; Hegg, R.; Manikhas, A.; Shtivelband, M.; Krivorotko, P.; Batista López, N.; Campone, M.; et al. A Randomized Adaptive Phase II/III Study of Buparlisib, a Pan-Class I PI3K Inhibitor, Combined with Paclitaxel for the Treatment of HER2- Advanced Breast Cancer (BELLE-4). Ann. Oncol. 2017, 28, 313–320. [Google Scholar] [CrossRef]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 Is a Highly Selective Intravenous PI3K Inhibitor with Potent P110α and P110δ Activities in Tumor Cell Lines and Xenograft Models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef] [Green Version]
- Commissioner, O. DA Approves New Treatment for Adults with Relapsed Follicular Lymphoma. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-follicular-lymphoma (accessed on 14 September 2017).
- National Cancer Institute (NCI). Phase Ib/II Trial of Copanlisib in Combination with Trastuzumab and Pertuzumab After Induction Treatment of HER2 Positive (HER2+) Metastatic Breast Cancer (MBC) with PIK3CA Mutation or PTEN Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT04108858 (accessed on 25 January 2022).
- Chauhan, A.F.; Cheson, B.D. Copanlisib in the Treatment of Relapsed Follicular Lymphoma: Utility and Experience from the Clinic. Cancer Manag. Res. 2021, 13, 677–692. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- FDA Approves Alpelisib for Metastatic Breast Cancer. FDA 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer (accessed on 24 May 2019).
- Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers 2021, 13, 3517. [Google Scholar] [CrossRef]
- Mavratzas, A.; Marmé, F. Alpelisib in the Treatment of Metastatic HR+ Breast Cancer with PIK3CA Mutations. Future Oncol. 2021, 17, 13–36. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor—Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor—Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Zumsteg, Z.S.; Morse, N.; Krigsfeld, G.; Gupta, G.; Higginson, D.S.; Lee, N.Y.; Morris, L.; Ganly, I.; Shiao, S.L.; Powell, S.N.; et al. Taselisib (GDC-0032), a Potent β-Sparing Small Molecule Inhibitor of PI3K, Radiosensitizes Head and Neck Squamous Carcinomas Containing Activating PIK3CA Alterations. Clin. Cancer Res. 2016, 22, 2009–2019. [Google Scholar] [CrossRef] [Green Version]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-Isopropyl-3-Methyl-1H-1,2–4-Triazol-5-Yl)-5,6-Dihydrobenzo[f]Imidazo[1,2-d][1,4]Oxazepin-9-Yl]-1H-Pyrazol-1-Yl}-2-Methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in vivo Antitumor Activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [CrossRef]
- Baselga, J.; Dent, S.F.; Cortés, J.; Im, Y.-H.; Diéras, V.; Harbeck, N.; Krop, I.E.; Verma, S.; Wilson, T.R.; Jin, H.; et al. Phase III Study of Taselisib (GDC-0032) + Fulvestrant (FULV) v FULV in Patients (pts) with Estrogen Receptor (ER)-Positive, PIK3CA-Mutant (MUT), Locally Advanced or Metastatic Breast Cancer (MBC): Primary Analysis from SANDPIPER. J. Clin. Oncol. 2018, 36, LBA1006. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Lara, P.N.; Longmate, J.; Mack, P.C.; Kelly, K.; Socinski, M.A.; Salgia, R.; Gitlitz, B.; Li, T.; Koczywas, M.; Reckamp, K.L.; et al. Phase II Study of the AKT Inhibitor MK-2206 plus Erlotinib in Patients with Advanced Non-Small Cell Lung Cancer Who Previously Progressed on Erlotinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.Y.; Walter, R.B.; Faderl, S.H.; Jabbour, E.J.; Zeng, Z.; Borthakur, G.; Huang, X.; Kadia, T.M.; Ruvolo, P.P.; Feliu, J.B.; et al. Preclinical and Early Clinical Evaluation of the Oral AKT Inhibitor, MK-2206, for the Treatment of Acute Myelogenous Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2226–2235. [Google Scholar] [CrossRef] [Green Version]
- Do, K.; Speranza, G.; Bishop, R.; Khin, S.; Rubinstein, L.; Kinders, R.J.; Datiles, M.I.B.; Eugeni, M.; Lam, M.H.; Doyle, L.A.; et al. Biomarker-Driven Phase 2 Study of MK-2206 and Selumetinib (AZD6244, ARRY-142886) in Patients with Colorectal Cancer. Investig. New Drugs 2015, 33, 720–728. [Google Scholar] [CrossRef]
- Ma, B.B.Y.; Lui, V.W.Y.; Hui, C.W.C.; Lau, C.P.Y.; Wong, C.-H.; Hui, E.P.; Ng, M.H.; Tsao, S.W.; Li, Y.; Chan, A.T.C. Preclinical evaluation of the AKT Inhibitor MK-2206 in Nasopharyngeal Carcinoma Cell Lines. Investig. New Drugs 2013, 31, 567–575. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, G.; Qiu, Z. Akt Inhibitor MK-2206 Reduces Pancreatic Cancer Cell Viability and Increases the Efficacy of Gemcitabine. Oncol. Lett. 2020, 19, 1999–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrikopoulou, A.; Chatzinikolaou, S.; Panourgias, E.; Kaparelou, M.; Liontos, M.; Dimopoulos, M.-A.; Zagouri, F. The Emerging Role of Capivasertib in Breast Cancer. Breast Off. J. Eur. Soc. Mastology 2022, 63, 157–167. [Google Scholar] [CrossRef] [PubMed]
- She, Q.-B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways That Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Shore, N.; Mellado, B.; Shah, S.; Hauke, R.; Costin, D.; Adra, N.; Cullberg, M.; Teruel, C.F.; Morris, T. A Phase I Study of Capivasertib in Combination with Abiraterone Acetate in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2022, 39, 85. [Google Scholar] [CrossRef]
- Li, J.; Davies, B.R.; Han, S.; Zhou, M.; Bai, Y.; Zhang, J.; Xu, Y.; Tang, L.; Wang, H.; Liu, Y.J.; et al. The AKT Inhibitor AZD5363 Is Selectively Active in PI3KCA Mutant Gastric Cancer, and Sensitizes a Patient-Derived Gastric Cancer Xenograft Model with PTEN Loss to Taxotere. J. Transl. Med. 2013, 11, 241. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, J.; Zhen, R.; Lv, J.; Zheng, L.; Su, X.; Zhu, G.; Gavine, P.R.; Xu, S.; Lu, S.; et al. Trastuzumab Anti-Tumor Efficacy in Patient-Derived Esophageal Squamous Cell Carcinoma Xenograft (PDECX) Mouse Models. J. Transl. Med. 2012, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, M.; Thavasu, P.; Stewart, A.; de Bono, J.; O’Brien, M.; Popat, S.; Bhosle, J.; Banerji, U. AKT Inhibition Synergistically Enhances Growth-Inhibitory Effects of Gefitinib and Increases Apoptosis in Non-Small Cell Lung Cancer Cell Lines. Lung Cancer 2014, 85, 141–146. [Google Scholar] [CrossRef]
- Smyth, L.M.; Tamura, K.; Oliveira, M.; Ciruelos, E.M.; Mayer, I.A.; Sablin, M.-P.; Biganzoli, L.; Ambrose, H.J.; Ashton, J.; Barnicle, A.; et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3947–3957. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.H.; Casbard, A.; Carucci, M.; Cox, C.; Butler, R.; Alchami, F.; Madden, T.-A.; Bale, C.; Bezecny, P.; Joffe, J.; et al. Fulvestrant plus Capivasertib versus Placebo after Relapse or Progression on an Aromatase Inhibitor in Metastatic, Oestrogen Receptor-Positive Breast Cancer (FAKTION): A Multicentre, Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2020, 21, 345–357. [Google Scholar] [CrossRef]
- Capivasertib Plus Faslodex Reduced the Risk of Disease Progression or Death by 40% versus Faslodex in Advanced HR-Positive Breast Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2022/capivasertib-pfs-in-hr-positive-breast-cancer.html (accessed on 8 December 2022).
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Bhattarai, T.S.; Shamu, T.; Gorelick, A.N.; Chang, M.T.; Chakravarty, D.; Gavrila, E.I.; Donoghue, M.T.A.; Gao, J.; Patel, S.; Gao, S.P.; et al. AKT Mutant Allele-Specific Activation Dictates Pharmacologic Sensitivities. Nat. Commun. 2022, 13, 2111. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-In-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, S.; Tabernero, J.; Molife, L.; Soria, J.-C.; Cervantes, A.; Vogelzang, N.; Patel, M.; Hussain, M.; Baron, A.; Argilés, G.; et al. Antitumor Activity of Ipatasertib Combined with Chemotherapy: Results from a Phase Ib Study in Solid Tumors. Ann. Oncol. 2020, 31, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-B.; Dent, R.; Im, S.-A.; Espié, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus Paclitaxel versus Placebo plus Paclitaxel as First-Line Therapy for Metastatic Triple-Negative Breast Cancer (LOTUS): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Turner, N.; Dent, R.A.; O’Shaughnessy, J.; Kim, S.-B.; Isakoff, S.J.; Barrios, C.; Saji, S.; Bondarenko, I.; Nowecki, Z.; Lian, Q.; et al. Ipatasertib plus Paclitaxel for PIK3CA/AKT1/PTEN-Altered Hormone Receptor-Positive HER2-Negative Advanced Breast Cancer: Primary Results from Cohort B of the IPATunity130 Randomized Phase 3 Trial. Breast Cancer Res. Treat. 2022, 191, 565–576. [Google Scholar] [CrossRef]
- Turner, N.C.; Alarcón, E.; Armstrong, A.C.; Philco, M.; Chuken, Y.L.; Sablin, M.-P.; Tamura, K.; Villanueva, A.G.; Pérez-Fidalgo, J.A.; Cheung, S.Y.A.; et al. BEECH: A Dose-Finding Run-In Followed by a Randomised Phase II Study Assessing the Efficacy of AKT Inhibitor Capivasertib (AZD5363) Combined with Paclitaxel in Patients with Estrogen Receptor-Positive Advanced or Metastatic Breast Cancer, and in a PIK3CA Mutant Sub-Population. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 774–780. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Mukhopadhyay, S.; Tung, K.; Patel, D.; Foster, D.A. Rapamycin-Induced G1 Cell Cycle Arrest Employs Both TGF-β and Rb Pathways. Cancer Lett. 2015, 360, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Yellen, P.; Chatterjee, A.; Preda, A.; Foster, D.A. Inhibition of S6 Kinase Suppresses the Apoptotic Effect of eIF4E Ablation by Inducing TGF-β-Dependent G1 Cell Cycle Arrest. Cancer Lett. 2013, 333, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellen, P.; Saqcena, M.; Salloum, D.; Feng, J.; Preda, A.; Xu, L.; Rodrik-Outmezguine, V.; Foster, D.A. High-Dose Rapamycin Induces Apoptosis in Human Cancer Cells by Dissociating mTOR Complex 1 and Suppressing Phosphorylation of 4E-BP1. Cell Cycle 2011, 10, 3948–3956. [Google Scholar] [CrossRef] [Green Version]
- MTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/16452206/ (accessed on 1 January 2023).
- Sun, S.-Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [Green Version]
- Le Gendre, O.; Sookdeo, A.; Duliepre, S.-A.; Utter, M.; Frias, M.; Foster, D.A. Suppression of AKT Phosphorylation Restores Rapamycin-Based Synthetic Lethality in SMAD4-Defective Pancreatic Cancer Cells. Mol. Cancer Res. 2013, 11, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-Cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The Dual Specificity PI3K/mTOR Inhibitor PKI-587 Displays Efficacy against T-Cell Acute Lymphoblastic Leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular Basis for Class Side Effects Associated with PI3K/AKT/mTOR Pathway Inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB Receptors: From Oncogenes to Targeted Cancer Therapies. Available online: https://www.jci.org/articles/view/32278/pdf (accessed on 13 November 2022).
- Pistilli, B.; Pluard, T.; Urruticoechea, A.; Farci, D.; Kong, A.; Bachelot, T.; Chan, S.; Han, H.S.; Jerusalem, G.; Urban, P.; et al. Phase II Study of Buparlisib (BKM120) and Trastuzumab in Patients with HER2+ Locally Advanced or Metastatic Breast Cancer Resistant to Trastuzumab-Based Therapy. Breast Cancer Res. Treat. 2018, 168, 357–364. [Google Scholar] [CrossRef]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; et al. Targeting Ligand-Activated ErbB2 Signaling Inhibits Breast and Prostate Tumor Growth. Cancer Cell 2002, 2, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, C.C.; Schuler, M.H.; Machiels, J.-P.H.; Hess, D.; Paz-Ares, L.; Awada, A.; von Moos, R.; Steeghs, N.; Ahnert, J.R.; De Mesmaeker, P.; et al. Phase Ib Study of Buparlisib (BKM120) plus Either Paclitaxel (PTX) in Advanced Solid Tumors (aST) or PTX plus Trastuzumab (TZ) in HER2+ Breast Cancer (BC). J. Clin. Oncol. 2014, 32, 627. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, C.R.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Pathways in Controlling Growth and Sensitivity to Therapy-Implications for Cancer and Aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Shapiro, G.I.; LoRusso, P.; Kwak, E.; Pandya, S.; Rudin, C.M.; Kurkjian, C.; Cleary, J.M.; Pilat, M.J.; Jones, S.; de Crespigny, A.; et al. Phase Ib Study of the MEK Inhibitor Cobimetinib (GDC-0973) in Combination with the PI3K Inhibitor Pictilisib (GDC-0941) in Patients with Advanced Solid Tumors. Investig. New Drugs 2020, 38, 419–432. [Google Scholar] [CrossRef]
- Britten, C.D. PI3K and MEK Inhibitor Combinations: Examining the Evidence in Selected Tumor Types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK Signaling Pathways Inhibitors as Anticancer Agents: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Merchant, M.; Orr, C.; Chan, J.; Otter, D.D.; Berry, L.; Kasman, I.; Koeppen, H.; Rice, K.; Yang, N.-Y.; et al. Intermittent Administration of MEK Inhibitor GDC-0973 plus PI3K Inhibitor GDC-0941 Triggers Robust Apoptosis and Tumor Growth Inhibition. Cancer Res. 2012, 72, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo Antitumor Activity of MEK and Phosphatidylinositol 3-Kinase Inhibitors in Basal-Like Breast Cancer Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A Phase Ib Dose-Escalation and Expansion Study of the Oral MEK Inhibitor Pimasertib and PI3K/MTOR Inhibitor Voxtalisib in Patients with Advanced Solid Tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algazi, A.P.; Rotow, J.; Posch, C.; Ortiz-Urda, S.; Pelayo, A.; Munster, P.N.; Daud, A. A Dual Pathway Inhibition Strategy Using BKM120 Combined with Vemurafenib Is Poorly Tolerated in BRAF V600 E/K Mutant Advanced Melanoma. Pigment. Cell Melanoma Res. 2019, 32, 603–606. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I Study of the MEK Inhibitor Trametinib in Combination with the AKT Inhibitor Afuresertib in Patients with Solid Tumors and Multiple Myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; A Gameiro, P.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras Decouples Glucose and Glutamine Metabolism to Support Cancer Cell Growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Kole, H.K.; Resnick, R.J.; Van Doren, M.; Racker, E. Regulation of 6-Phosphofructo-1-Kinase Activity in Ras-Transformed Rat-1 Fibroblasts. Arch. Biochem. Biophys. 1991, 286, 586–590. [Google Scholar] [CrossRef]
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and Methylaminoisobutyrate Uptake in Rat-1 Cells Transfected with Ras or Myc Oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538. [Google Scholar] [CrossRef]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS Modulates Mitochondrial Metabolism in Human Colon Cancer Cells by Inducing HIF-1α and HIF-2α Target Genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Opel, D.; Naumann, I.; Kappler, R.; Friedman, L.; von Schweinitz, D.; Debatin, K.-M.; Fulda, S. PI3K Inhibitors Prime Neuroblastoma Cells for Chemotherapy by Shifting the Balance towards Pro-Apoptotic Bcl-2 Proteins and Enhanced Mitochondrial Apoptosis. Oncogene 2011, 30, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Opel, D.; Naumann, I.; Schneider, M.; Bertele, D.; Debatin, K.-M.; Fulda, S. Targeting Aberrant PI3K/Akt Activation by PI103 Restores Sensitivity to TRAIL-Induced Apoptosis in Neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3233–3247. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Fujiwara, Y.; Tamura, K.; Kondo, S.; Iwasa, S.; Tanabe, Y.; Horiike, A.; Yanagitani, N.; Kitazono, S.; Inatani, M.; et al. Phase Ia/Ib Study of the Pan-Class I PI3K Inhibitor Pictilisib (GDC-0941) Administered as a Single Agent in Japanese Patients with Solid Tumors and in Combination in Japanese Patients with Non-Squamous Non-Small Cell Lung Cancer. Investig. New Drugs 2017, 35, 37–46. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kang, Y.-K.; Ng, M.; Chung, H.; Wainberg, Z.; Gendreau, S.; Chan, W.; Xu, N.; Maslyar, D.; Meng, R.; et al. A Phase II, Randomised Study of mFOLFOX6 with or without the Akt Inhibitor Ipatasertib in Patients with Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer. Eur. J. Cancer 2019, 108, 17–24. [Google Scholar] [CrossRef]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear Phosphoinositide 3-Kinase β Controls Double-Strand Break DNA Repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [Green Version]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of Phosphatidylinositol-3-OH Kinase/Akt Signaling Impairs DNA Repair in Glioblastoma Cells following Ionizing Radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP Is Effective in the Treatment of Ovarian Cancer Cells with Wild-Type PIK3CA Genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batalini, F.; Xiong, N.; Tayob, N.; Polak, M.; Eismann, J.; Cantley, L.C.; Shapiro, G.I.; Adalsteinsson, V.; Winer, E.P.; Konstantinopoulos, P.A.; et al. Phase 1b Clinical Trial with Alpelisib plus Olaparib for Patients with Advanced Triple-Negative Breast Cancer. Clin. Cancer Res. 2022, 28, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.; Wulf, G.M.; Birrer, M.J.; Westin, S.N.; Quy, P.; Bell-McGuinn, K.M.; Lasonde, B.; Whalen, C.; Aghajanian, C.; Solit, D.B.; et al. Phase I Study of Oral BKM120 and Oral Olaparib for High-Grade Serous Ovarian Cancer (HGSC) or Triple-negative Breast Cancer (TNBC). J. Clin. Oncol. 2014, 32, 2510. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN Loss Increases PD-L1 Protein Expression and Affects the Correlation between PD-L1 Expression and Clinical Parameters in Colorectal Cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression That Is Reversible by MEK and PI3K Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K Orchestration of the in vivo Persistence of Chimeric Antigen Receptor-Modified T Cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Perkins, D.M.R.; Grande, S.; Hamel, B.A.; Horton, H.M.; Garrett, B.T.E.; Miller, S.M.; Latimer, I.H.J.; Horvath, D.C.J.; Kuczewski, M.M.; Friedman, K.M.; et al. Manufacturing an Enhanced CAR T Cell Product By Inhibition of the PI3K/Akt Pathway during T Cell Expansion Results in Improved in vivo Efficacy of Anti-BCMA CAR T Cells. Blood 2015, 126, 1893. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Schrijver, W.A.M.E.; Suijkerbuijk, K.P.M.; van Gils, C.H.; van der Wall, E.; Moelans, C.B.; van Diest, P.J. Receptor Conversion in Distant Breast Cancer Metastases: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2018, 110, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Thulin, A.; Andersson, C.; Rönnerman, E.W.; De Lara, S.; Chamalidou, C.; Schoenfeld, A.; Kovács, A.; Fagman, H.; Enlund, F.; Linderholm, B.K. Discordance of PIK3CA and TP53 Mutations between Breast Cancer Brain Metastases and Matched Primary Tumors. Sci. Rep. 2021, 11, 23548. [Google Scholar] [CrossRef]
- Fumagalli, C.; Ranghiero, A.; Gandini, S.; Corso, F.; Taormina, S.; De Camilli, E.; Rappa, A.; Vacirca, D.; Viale, G.; Guerini-Rocco, E.; et al. Inter-Tumor Genomic Heterogeneity of Breast Cancers: Comprehensive Genomic Profile of Primary Early Breast Cancers and Relapses. Breast Cancer Res. BCR 2020, 22, 107. [Google Scholar] [CrossRef]
- Toppmeyer, D.L.; Press, M.F. Testing Considerations for Phosphatidylinositol-3-Kinase Catalytic Subunit Alpha as an Emerging Biomarker in Advanced Breast Cancer. Cancer Med. 2020, 9, 6463–6472. [Google Scholar] [CrossRef]
- Dumbrava, E.; Call, S.; Huang, H.; Stuckett, A.; Madwani, K.; Adat, A.; Hong, D.; Piha-Paul, S.; Subbiah, V.; Karp, D.; et al. PIK3CA Mutations in Plasma Circulating Tumor DNA Predict Survival and Treatment Outcomes in Patients with Advanced Cancers. ESMO Open 2021, 6, 100230. [Google Scholar] [CrossRef]
- Brandão, M.; Caparica, R.; Eiger, D.; de Azambuja, E. Biomarkers of Response and Resistance to PI3K Inhibitors in Estrogen Receptor-Positive Breast Cancer Patients and Combination Therapies Involving PI3K Inhibitors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, x27–x42. [Google Scholar] [CrossRef] [Green Version]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; Debeaumont, R.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-Deficient Cancers Depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.; Wallin, J.J.; Sampath, D.; GuhaThakurta, D.; Savage, H.; Punnoose, E.A.; Guan, J.; Berry, L.; Prior, W.W.; Amler, L.C.; et al. Predictive Biomarkers of Sensitivity to the Phosphatidylinositol 3′ Kinase Inhibitor GDC-0941 in Breast Cancer Preclinical Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3670–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of Insulin Feedback Enhances the Efficacy of PI3K Inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K Inhibition Results in Enhanced Estrogen Receptor Function and Dependence in Hormone Receptor—Positive Breast Cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E.; Juric, D.; Solit, D.; Berger, M.F.; Won, H.H.; et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [Green Version]
- El Bairi, K.; Haynes, H.R.; Blackley, E.; Fineberg, S.; Shear, J.; Turner, S.; de Freitas, J.R.; Sur, D.; Amendola, L.C.; Gharib, M.; et al. The Tale of TILs in Breast Cancer: A Report from The International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 2021, 7, 150. [Google Scholar] [CrossRef]
- Gagliato, D.D.M.; Cortes, J.; Curigliano, G.; Loi, S.; Denkert, C.; Perez-Garcia, J.; Holgado, E. Tumor-Infiltrating Lymphocytes in Breast Cancer and Implications for Clinical Practice. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 527–537. [Google Scholar] [CrossRef]
- Mego, M.; Gao, H.; Cohen, E.; Anfossi, S.; Giordano, A.; Sanda, T.; Fouad, T.; De Giorgi, U.; Giuliano, M.; Woodward, W.; et al. Circulating Tumor Cells (CTC) Are Associated with Defects in Adaptive Immunity in Patients with Inflammatory Breast Cancer. J. Cancer 2016, 7, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- De Giorgi, U.; Mego, M.; Scarpi, E.; Giordano, A.; Giuliano, M.; Valero, V.; Alvarez, R.H.; Ueno, N.T.; Cristofanilli, M.; Reuben, J.M. Association between Circulating Tumor Cells and Peripheral Blood Monocytes in Metastatic Breast Cancer. Ther. Adv. Med Oncol. 2019, 11, 1758835919866065. [Google Scholar] [CrossRef] [Green Version]
- Gianni, C.; Palleschi, M.; Schepisi, G.; Casadei, C.; Bleve, S.; Merloni, F.; Sirico, M.; Sarti, S.; Cecconetto, L.; Di Menna, G.; et al. Circulating Inflammatory Cells in Patients with Metastatic Breast Cancer: Implications for Treatment. Front. Oncol. 2022, 12, 882896. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial | Study Design | Intervention | Settings | Primary Endpoint | Phase | Status |
|---|---|---|---|---|---|---|
| NCT04975958 | 63 Participants Interventional Non-Randomized Parallel Assignment Open Label | Buparlisib Atezolizumab AN0025 | Advanced Solid tumors | DLTs | 1 | Recruiting |
| NCT04338399 (BURAN) | 483 Participants Interventional Randomized Parallel Assignment Open Label | Buparlisib Paclitaxel | mHNCC | OS | 3 | Recruiting |
| NCT04108858 | 12 Participants Interventional Parallel Assignment Open Label | Copanlisib Pertuzumab Trastuzumab | PI3KCA/PTEN mutated HER2+/HR- MBC | AEs | 1/2 | Recruiting |
| NCT04572763 | 48 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Copanlisib Venetoclax | Relapsed/refractory DLBCL | MTD, ORR | 1/2 | Active Not recruiting |
| NCT03711058 | 18 Participants Interventional Non-Randomized Sequential Assignment Open Label | Copanlisib Nivolumab | MSS CRC | MTD, DLT | 1/2 | Active Not recruiting |
| NCT04253262 | 13 Participants Interventional Non-Randomized Sequential Assignment Open Label | Copanlisib Rucaparib | mCRPC | MTD | 1/2 | Active Not recruiting |
| NCT03502733 | 48 Participants Interventional Single-Group Assignment Open Label | Copanlisib Ipililumab Nivolumab | Advanced cancer, Lymphoma | RP2D | 1 | Active, Not recruiting |
| NCT03484819 | 106 Participants Interventional Single-Group Assignment Open Label | Copanlisib Hydrochlorid Nivolumab | Refractory DLBCL PMBCL | ORR | 2 | Active, Not recruiting |
| NCT02367040 CHRONOS-3 | 458 Participants Interventional Randomized Parallel Assignment | Copanlisib Rituximab | Relapsed iNHL | PFS | 2 | Active, Not recruiting |
| NCT01660451 | 227 Participants Interventional Non-Randomized Parallel Assignment Open Label | Copanlisib | Indolent or aggressive NHL | ORR | Active, Not recruiting | |
| NCT05143229 | 18 Participants Interventional Non-Randomized Sequential Assignment Open Label | Alpelisib Sacituzumab Govitecan | Stage III/Stage IV HR+/HER2− MBC | RP2D | 1 | Recruiting |
| NCT04208178 (EPIK-B2) | 551 Participants Interventional Randomized Parallel Assignment | Alpelisib Trastuzumab Pertuzumab | PIK3CA mutated HER2+ MBC | PFS | 3 | Recruiting |
| NCT04762979 | 44 Participants Interventional Single-Group Assignment Open Label | Alpelisib Fulvestrant Aromatase inhibitor | PIK3CA mutated HR+/HER2− MBC | PFS | 2 | Recruiting |
| NCT05508906 | 60 Participants Interventional Non-Randomized Parallel Assignment Open Label | Alpelisib Ribociclib OP-1250 | HR+/HER2− MBC | DLTsMTD | 1 | Recruiting |
| NCT04251533 | 566 Participants Interventional Randomized Parallel Assignment | Alpelisib Nab paclitaxel Placebo | PIK3CA mutated/PTEN loss mTNBC | PFS, ORR | 3 | Recruiting |
| NCT05025735 | 25 Participants Interventional Randomized Single-Group Assignment Open Label | Alpelisib Dapagliflozin Fulvestrant | PI3KCA mutated HR+/HER2− MBC | Incidence of all grade hyperglycemia | 2 | Recruiting |
| NCT05230810 | 40 Participants Interventional Single-Group Assignment Open Label | Alpelisib Fulvestrant Tucatinib | PIK3CA mutated HER2+ MBC | AEs | 1/2 | Recruiting |
| NCT05501886 (VIKTORIA-1) | 701 Participants Interventional Randomized Parallel Assignment Open Label | Alpelisib Gedatolisib Palbociclib Fulvestrant | HR+/HER2− MBC | PFS | 3 | Recruiting |
| NCT04997902 | 36 Participants Interventional Parallel Assignment Open Label | Alpelisib Tipifarnib | mHNCC | DLTs | 1/2 | Recruiting |
| NCT05063786 | 358 Participants Interventional Single-Group Assignment Open Label | Alpelisib Olaparib Paclitaxel PLD | metastatic OC | PFS | 3 | Recruiting |
| NCT04526470 | 55 Participants Interventional Single-Group Assignment Open Label | Alpelisib Paclitaxel | PIK3CA mutated GA | MTD RP2D | 1/2 | Recruiting |
| NCT03207529 | 28 Participants Interventional Single-Group Assignment Open Label | Alpelisib Enzalutamide | AR+/PTEN positive MBC | MTD | 1 | Recruiting |
| NCT01872260 | 255 Participants Interventional Randomized Parallel Assignment Open Label | Alpelisib Letrozole LEE011 | HR+/HER2− MBC | DLTs Safety | 1/2 | Active, Not yet recruiting |
| NCT03284957 (AMEERA-1) | 136 Participants Interventional Randomized Parallel Assignment Open Label | Amcenestrant Palbociclib Alpelisib Everolimus Abemaciclib | HR+/HER2− MBC | DLTs | Active, Not yet recruiting | |
| NCT04666038 (BRUIN CLL-321) | 250 Participants Interventional Randomized Parallel Assignment Open Label | Idelalisib LOXO-305 Bendamustine Rituximab | Chronic CLL/SLL | PFS | 3 | Recruiting |
| NCT03890289 (GAUDEALIS) | 5 Participants Interventional Single-Group Assignment Open Label | Idelalisib Obinutuzumab | Refractory FL | ORR | 2 | Active Not yet recruiting |
| NCT02787369 | 3 Participants Interventional Non-Randomized Parallel Assignment Open Label | Idelalisib ACY-1215 Ibrutinib | Refractory CLL | MTD | 1 | Active Not yet recruiting |
| NCT02970318 | 311 Participants Interventional Randomized Parallel Assignment | Idelalisib calabrutinib (ACP-196) Rituximab Bendamustine | Refractory CLL | PFS | 3 | Active Not yet recruiting |
| NCT02135133 | 50 Participants Interventional Single-Group Assignment Open Label | Idelalisib Ofatumumab | CLL/SLL | ORR | 2 | Active, Not recruiting |
| NCT04191499 | 400 Participants Interventional Randomized Parallel Assignment | Inavolisib Palbociclib Fulvestrant | PIK3CA mutated HR+/HER2+ MBC | PFS | 2/3 | Recruiting |
| Clinical Trial | Study Design | Intervention | Settings | Primary Endpoint | Phase | Status |
|---|---|---|---|---|---|---|
| NCT03310541 | 12 Participants Interventional Parallel Assignment Open Label | Capivasertib, Enzalutamide, Fulvestrant | Advanced solid tumors harboring mutations in AKT1, AKT2, or AKT3 | ORR | 1 | Active, not yet recruiting |
| NCT05593497 (SNARE) | 30 Participants Interventional Single-Group Assignment Open Label | Capivasertib, Abiraterone Acetate Leuprolide | PTEN loss High-risk localized PC | pCR MRD | 2 | Not recruiting |
| NCT04439123 (MATCH-Subprotocol Y) | 35 Participants Interventional Single-Group Assignment Open Label | Capivasertib | Cancers with AKT genetic changes | ORR | 2 | Active, not yet recruiting |
| NCT04851613 | 20 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Afuresertib, Fulvestrant | Locally advanced or HR+/HER2− MBC | ORR | 1 | Recruiting |
| NCT04374630 (PROFECTA-II) | 141 Participants Interventional Parallel Assisgnment Open Label | Afuresertib Paclitaxel | Platinum-resistant ovarian cancer | rPFS | 2 | Recruiting |
| NCT05383482 | 167 Participants Non-Randomized Sequential Assignment Open Label | Afuresertib Nab paclitaxel Docetaxel Sintilimab | Solid tumors Resistant to prior anti-PD-1/PD-L1 | AEs DLTs | 1/2 | Recruiting |
| NCT05390710 | 101 Participants Randomized Sequential Assignment Open Label | LAE005 + Afuresertib Nab-Paclitaxel | Metastatic TNBC | AEs DLT | 1/2 | Recruiting |
| NCT04060394 | 74 Participants Randomized Sequential Assignment Open Label | Afuresertib LAE001/prednisone + | mCRPC | rPFS | 1/2 | Recruiting |
| NCT04253561 (IPATHER) | 25 Participants Interventional Single-Group Assignment Open Label | Ipatasertib Trastuzumab Pertuzumab | HER2+ PI3KCA mutant MBC | RP2D | 1 | Recruiting |
| NCT05172245 | 36 Participants Interventional Single-Group Assignment Open Label | Ipatasertib Cisplatin Radiation Therapy | Stage III-IVB HNC | MTD RP2D | 1 | Recruiting |
| NCT04467801 (Ipat-Lung) | 60 Participants Interventional Single-Group Assignment Open Label | Ipatasertib Docetaxel | mNSCLC | PFS | 2 | Recruiting |
| NCT03673787 | 87 Participants Interventional Non-Randomized Parallel Assignment Open Label | Ipatasertib atezolizumab | Glioblastoma Multiforme mPC | MTD | 1/2 | Recruiting |
| NCT05276973 | 24 Participants Interventional Single-Group Assignment Open Label | Ipatasertib Carboplatin Paclitaxel | Stage III or IV Epithelial OC | MTD | 1 | Recruiting |
| NCT03959891 (TAKTIC) | 60 Participants Interventional Non-Randomized Parallel Assignment Open Label | Ipatasertib Fulvestrant Aromatase Inhibitor Palbociclib | HR+/HER2− mBC | TEAE | 1 | Recruiting |
| NCT04650581 (FINER) | 250 Participants Interventional Randomized Parallel Assignment | Ipatasertib Fulvestrant | HR+/HER2− mBC | PFS | 3 | Recruiting |
| NCT04920708 Without ctDNA Suppression (FAIM) | 324 Participants Interventional Randomized Parallel Assignment Open Label | Ipatasertib Fulvestrant Palbociclib | HR+/HER2− mBC | PFS | 2 | Not yet recruiting |
| NCT04464174 (PATHFINDER) | 54 Participants Interventional Non-randomized Parallel Assignment Open Label | Ipatasertib non-taxane chemotherapy | mTNBC | Safety | 2 | Active, not yet recruiting |
| NCT03853707 | 28 Participants Interventional Randomized Parallel Assignment Open Label | Ipatasertib Atezolizumab Capecitabine Carboplatin Ipatasertib Paclitaxel | mTNBC | RP2D, PFS | 1/2 | Active, not yet recruiting |
| NCT05498896 (BARBICAN) | 146 Participants Interventional Non-randomized Parallel Assignment Open Label | Ipatasertib Atezolizumab Paclitaxel Doxorubicin Cyclophosphamide | mTNBC | pCR | 2 | Active, not yet recruiting |
| NCT03072238 (IPATential150) | 1101 Participants Interventional Parallel Assignment | Ipatasertib Abiraterone Placebo | mCRPC | rPFS | 3 | Active, not yet recruiting |
| NCT05538897 | 96 Participants Interventional Randomized Parallel Assignment Open Label | Ipatasertib Megestrol Acetate | mEC | AEs | 1/2 | Not yet recruiting |
| NCT04739202 ((IMMUNOGAST) | 60 Participants Interventional Non-Randomized Parallel Assignment Open Label | Ipatasertib Atezolizumab | mGA | ORR | 2 | Recruiting |
| Clinical Trial | Study Design | Intervention | Settings | Primary Endpoint | Phase | Status |
|---|---|---|---|---|---|---|
| NCT03698383 | 15 Participants Interventional Single-Group Assignment Open Label | Trastuzumab biosimilars (Herzuma) Gedatolisib | HER2+ MBC | ORR | 2 | Recruiting |
| NCT03911973 | 52 Participants Interventional Single-Group Assignment Open Label | Talazoparib Gedatolisib | mTNBC | ORR | ½ | Recruiting |
| NCT03065062 | 96 Participants Interventional Single-Group Assignment Open Label | Palbociclib Gedatolisib | Solid tumors | MTD, RP2D | 1 | Recruiting |
| NCT05501886 (VIKTORIA-1) | 141 Participants Interventional Randomized Parallel Assisgnment Open Label | Palbociclib Fulvestrant Alpelisib Gedatolisib | HR+/HER2 MBC | PFS | 3 | Recruiting |
| Clinical Trial | Study Design | Intervention | Settings | Primary Endpoint | Phase | Status |
|---|---|---|---|---|---|---|
| NCT04431635 | 35 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Copanlisib Nivolumab Rituximab | Relapsed/refractory indolent follicular or marginal zone lymphoma | MDT CR rate | Ib | Recruiting |
| NCT03961698 | 91 Participants Interventional Non-Randomized Parallel Assignment | Eganelisib Atezolizumab Nab-paclitaxel Bevacizumab | Metastatic TNBC or advanced RCC | CR rate | II | Active, not recruiting |
| NCT04317105 | 102 Participants Interventional Non-Randomized Parallel Assignment Open Label | Copanlisib Ipilimumab Nivolumab | Advanced malignant solid neoplasm | AEs, DLT | I/II | Recruiting |
| NCT03131908 | 36 Participants Interventional Non-Randomized Parallel Assignment Open Label | GSK2636771 Pembrolizumab | Metastatic PTEN loss melanoma | MTD ORR | I/II | Active, not recruiting |
| NCT03772561 | 40 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Capivasertib Olaparib Durvalumab | Advanced or metastatic solid tumor malignancies | ORR | I | Recruiting |
| NCT05387616 | 98 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Copanlisib Obinutuzumab | Follicular lymphoma | PFS | II | Recruiting |
| NCT03711058 | 54 Participants Interventional Non-Randomized Sequential Assignment Open Label | Copanlisib Nivolumab | MSS relapsed/refractory solid tumors and CRC | DLTs, ORR | I/II | Active, not recruiting |
| NCT03673787 | 87 Participants Non-Randomized Parallel Assignment Open Label | Ipatasertib Atezolizumab | Advanced solid tumours with PI3K pathway hyperactivation | MTD, AEs | I/II | Recruiting |
| NCT02637531 | 219 Participants Interventional Non-Randomized Single-Group Assignment Open Label | Eganelisib Nivolumab | Advanced solid tumours | DLTs, AEs | I/Ib | Active, not recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirico, M.; D’Angelo, A.; Gianni, C.; Casadei, C.; Merloni, F.; De Giorgi, U. Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy. Cancers 2023, 15, 703. https://doi.org/10.3390/cancers15030703
Sirico M, D’Angelo A, Gianni C, Casadei C, Merloni F, De Giorgi U. Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy. Cancers. 2023; 15(3):703. https://doi.org/10.3390/cancers15030703
Chicago/Turabian StyleSirico, Marianna, Alberto D’Angelo, Caterina Gianni, Chiara Casadei, Filippo Merloni, and Ugo De Giorgi. 2023. "Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy" Cancers 15, no. 3: 703. https://doi.org/10.3390/cancers15030703
APA StyleSirico, M., D’Angelo, A., Gianni, C., Casadei, C., Merloni, F., & De Giorgi, U. (2023). Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy. Cancers, 15(3), 703. https://doi.org/10.3390/cancers15030703