1. Introduction
Over the past years, despite the coronavirus disease (COVID-19) pandemic, the prevalence of breast cancer cases has raised dramatically and is ranked as the 2nd major cause of cancer-related death worldwide [
1]. About 287,850 new breast cancer cases with more than 43,250 breast cancer-related deaths is expecting in USA in 2022 [
1]. “Breast cancer is ranked as the first leading cause of cancer-related deaths in Saudi Arabia”. In 2020, the WHO estimated more than 3954 new breast cancer cases with over 1095 breast cancer-related deaths [
2,
3]. It has been revealed a substantial discrepancy in breast cancer incidence between Saudis and non-Saudis, which is likely due to gene expression and lifestyle differences [
4,
5]. Breast cancer is distinguished by its unique genetic, molecular, and clinical heterogeneity [
6,
7]. Recently molecular profiling has determined that 26% of hereditary tumors are caused by mutations in highly penetrant
genes (
BRCA2, PTEN, BRCA1, TP53, CDH1), which carry a lifelong risk of about 80% for breast cancer development [
8].
The
PI3K-AKT-mTOR signaling pathway plays a significant role in the susceptibility of breast carcinoma [
9]. Several
PI3K-AKT-mTOR inhibitors have been developed for targeting the components of PI3K-AKT-mTOR pathway and promising therapeutic effects has been reported in breast cancer [
10]. Some combination therapy with mTOR inhibitor and exemestane has significantly prolonged the overall survival (OS) of breast cancer patients with hormone receptor (HR)-positive metastatic breast cancer [
11]. Therefore, targeting the three components of
PI3K-AKT-mTOR signaling pathway has become promising therapeutic modalities in the treatment of breast cancer. With the advent of novel promising
PI3K-AKT-mTOR pathway inhibitors, it is essential to identify the specific patients who can benefit from these
PI3K-AKT-mTOR inhibitors. Next-generation sequencing (NGS) technology has made it possible to identify such genetic aberrations quickly and accurately. Many research studies have reported that breast cancer patients with
PIK3CA mutations (
Figure 1) have a better prognosis [
12], while Sobhani et al. reported a worse prognosis for women with
PIK3CA-mutant breast tumors [
13]. Gerratana et al. studied 88 cases of metastatic breast cancer by multivariate logistic regression and reported that the detection of
PIK3CA mutation in circulating tumor DNA was associated with lung metastasis (OR 3.74) [
14].
MDM4 (
MDM4 Regulator of P53) is a Protein Coding gene. [
15]. The human MDM4 gene, which plays a role in apoptosis, encodes a 490-amino acid protein containing a RING finger domain and a putative nuclear localization signal. These oncogenic capabilities are regularly accomplished through
MDM4 gene amplification. However, several mutations in
MDM4 were reported to be associated with cancers such as breast cancer, endometrial cancer, and stomach cancer [
16]. In breast malignancy, several SNPs, and mutations in
MDM4 were reported to promote breast growth including the rs11801299 G > A polymorphisms.
MDM4 rs11801299 G > A polymorphisms has been reported to be connected with retinoblastoma susceptibility [
17], risk of gastric cancer in Chinese population [
18], and risk of breast cancer [
19]. In 2018, an Iranian study reported an association between the
MDM4 rs11801299 G > A polymorphisms and the susceptibility to breast tumor [
20].
The Krüppel-like transcription factors (KLF) are a family of 17 transcription factors which are involved in the modulation of several genes that are essential for different cellular processes [
21]. Recently, the importance of
KLF14, also called
BTEB5, was postulated in different genetic studies revealing its significant role as a primary gene expression regulator [
22].
KLF 14 has been shown to be a novel tumour suppressor and is often downregulated in human cancers, demonstrating its role as an important biomarker for disease progression and for developing new cancers treatments [
23,
24]. Meta-analysis and genome wide studies examining the effect of polymorphic variants found that rs972283 polymorphism in
KLF14 has high risk of developing diseases with the G allele associated with
T2D, metabolic disorders in different populations and in another study with A allele which is associated with polycystic ovary syndrome in specific populations [
25,
26,
27]. For
T2D and breast cancer susceptible groups with the
KLF-14- rs972283 variant association found to be weak among European and African population [
28]. However, this variant is highly attributed to patient with genetic predisposition for T2D and with breast or prostate cancer [
29]. Breast cancer frequently experiences oncogenic activation of the phosphatidylinositol-3-kinase (PI3K), protein kinase B (PKB/AKT), and mammalian target of rapamycin (mTOR) pathway, which promotes tumor development, disease progression, and therapy resistance. Recent studies suggest that the intricate interactions between the
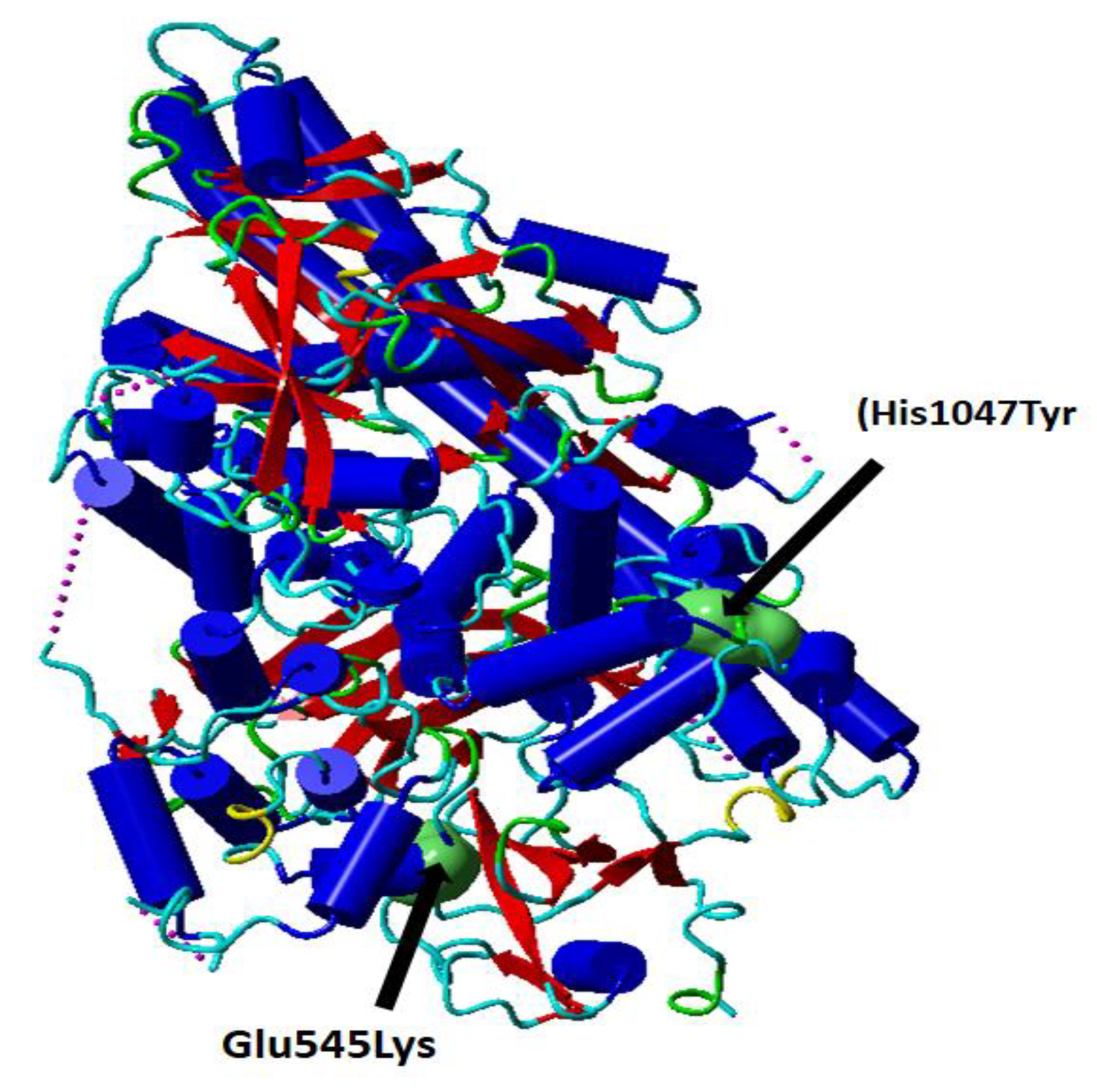
PI3K-AKT-mTOR pathway and several interacting cell signaling cascades can enhance the advancement of Breast cancer. The PIK3R1 gene encodes the PIK3R1 protein. PI3K is an important protein in the Akt signaling pathway which play important role in cell survival, differentiation, growth, glucose trafficking, and utilization. Glu545Lys (rs104886003); His1047Tyr (rs121913281) mutations induce confirmation change in the phosphatidylinositol- 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) protein (
Figure 1).
Figure 1.
Cartoon presentation of the catalytic subunit of PI3K secondary structure PDB ID: 5XGH. The single nucleotide variations rs104886003 G > A (Glu545Lys) and rs121913281 C > T (His1047Tyr) are shown in green surface presentation. This figure (Figure 1) is prepared using YASARA, and modified from Elfaki et al. [
30].
Figure 1.
Cartoon presentation of the catalytic subunit of PI3K secondary structure PDB ID: 5XGH. The single nucleotide variations rs104886003 G > A (Glu545Lys) and rs121913281 C > T (His1047Tyr) are shown in green surface presentation. This figure (Figure 1) is prepared using YASARA, and modified from Elfaki et al. [
30].
MicroRNAs are small, single stranded, non-coding RNAs. Most of them are down expressed in cancer cells which guides to cellular transformation followed by tumor development and cancer progression. The miR-27a-3p is a single stranded, non-coding RNAs which is recognized as an oncogenic RNA in multiple malignancies including colorectal, Breast and gastric cancer. MiR-27a is mostly located in exosomes of the BC cancer cells [
30,
31,
32]. It is reported that miR-27a-3p (exosomal) foster immune attach by activating PD-L1 via
MAGI2/PTEN/PI3K axis in breast cancer [
33]. The miR-27a rs895819 A/G is a common SNP that is found in the loop of the pre-miRNA and may have a key role in mi-R27a maturation [
34,
35]. The minimum free energy (MFE) is affected when there is a variation from A to G in this SNP, and as a result, the function of the miRNA might be affected to some extent. Several reported studies showed the possibility of this variant allele in decreasing the risk of cancer, while however other studies showed the opposite [
36]. In one of the meta-analysis, miR-27a rs895819 gene polymorphism has been reported to be linked with the breast cancer susceptibility among Caucasians. Besides AA genotype of miR-27a conferred strong link with breast cancer susceptibility and AG genotype (heterozygous) as well as G-allele were protective factors [
37].
Recent studies have shown that miR-196b acts as a tumor suppressor in various cancer types [
38] in HepG2 cells that suppressed cell proliferation and induced apoptosis [
39]. One of the most common SNP reported is
miR-196a-rs11614913 C to T which may increase or decrease the extent of translation of the target protein therefore may alter the expression as well as the functions which in turn can increase cancer susceptibility [
40]. MiR-196a has been reported to target several genes that may be involved in cell cycle, apoptosis, and differentiation.
MiR-196a targets annexin-
A1 (
ANXA1) gene which controls the physiological mechanisms such as exocytosis, hormone secretion, apoptosis, and signal transduction [
41]. Some studies have been performed to establish the link between
miR-196a (rs11614913) polymorphism and breast cancer risk [
42]. Some studies report it as a protective factor [
43] whereas others like
Omrani et al. and Qi et al. indicated strong association with the breast cancer susceptibility [
44,
45].
The human genome’s protein-coding regions, which contain around 85% of the disease-causing variations, can almost entirely be covered by whole-exome sequencing (WES) [
46]. The exome makes up around 1% of the entire human genome, making WES an incredibly potent tool for medical genetic research. Future case/control and family based NGS research will be more effective thanks to WES, which is frequently more affordable and permits the sequencing of more individuals [
47]. More recently, 65 loci strongly linked with breast cancer were found by a genome-wide association study [
48].
2. Materials and Methods
This study was conducted on primary BC patients (n = 115) and gender matched healthy women (n = 115) with no history of any type of cancer and who were not related to the patients from the general population. Formalin fixed paraffin embedded (FFPE) tissue specimens were obtained from the Division of Histopathology, King Salman Armed Forces Hospital North-western Region at Tabuk city and other hospitals. This study handled patient samples and records in accordance with the Declaration of Helsinki revised in 2013 under the ethical approval of Armed Forces Hospital Research Ethics Committee (KSAFH-REC-2020-345/8 September 2020) and the ethics committee of the University of Tabuk (protocol code UT-115-13-2020/24 April 2020). All patients obtained written informed consent form. The study experiment was carried out at Genome and Biotechnology Unit, Faculty of Science, University of Tabuk.
Inclusion criteria of patients:
The study included clinically confirmed cases of breast cancer patients who were Saudi women. Specimens were collected from primary breast cancer patients who had been diagnosed based on the clinical, histopathological, and radiological findings. The study also included breast cancer cases who had received chemotherapy, hormonal therapy, and radiotherapy.
Exclusion criteria:
The exclusion criteria included (1). Breast cancer patients diagnosed with multiple cancer types, (2). Patients who were unable to cope with the study protocol, (3). Non-Saudi women with breast cancer, (4). Any breast cancer patient with a history of previous significant malignancy.
Inclusion criteria for healthy controls
A healthy control cohort was prepared from the participants visiting for routine checkup to King Fahd Special Hospital, Tabuk, Saudi Arabia. These participants completed the informed consent form and filled questionnaire. The inclusion criteria included (1) Gender matched healthy women, (2) Women with age equal or greater than 40 years (≥40), (3) Only ethnic Saudi women.
Exclusion criteria for healthy controls
The exclusion criteria included healthy women with a family history of breast cancer, non-ethnic Saudi women, women with age below 40 years of age.
2.1. Demographic Data
Each breast cancer patient filled out a standardized questionnaire about their demographics, family history, and previous knowledge. To determine relevant clinical history, detailed laboratory and clinical data were collected.
2.2. DNA Extraction from Cases and Controls
The FFPE samples were collected from the pathology department and some peripheral blood specimens were also obtained by venipuncture and placed in EDTA tubes after assessing the clinicopathological findings. According to the manufacturer’s instructions, DNA was extracted from the specimens (FFPE) using the QIAamp DNA FFPE Tissue Kit (Cat-56404) from Qiagen (Hilden, Germany). Similarly, blood specimens were processed for genomic DNA extraction using the DNeasy Blood Kit (Cat-Noc69506) from Qiagen (Germany). The extracted DNA was dissolved in water devoid of nucleases and kept at 4 °C until needed.
The genomic DNA quality and integrity were checked using NanoDropTM (Thermo Scientific, Waltham, MA, USA), and the extracted DNA’s quality was investigated optically using the A260 nm/A280 nm ratio (1.83–1.99).
Genotyping of PI3K rs121913281 C > T, MDM4 rs11801299 A > G, AKT-1 rs1130233 G > A, KLF-14, MDM4, miRNAs 27a and miR-196a
The genotyping of the PI3K rs121913281 C > T, AKT-1 rs1130233 G > A, KLF 14-rs972283C > T, MDM4-rs11801299 A > G, miRNAs 27a -rs895819A > G and miR-196a-2 -rs11614913 C > T genes were determined using ARMS primers that were used elsewhere [
20,
26]. The ARMS primers for KLF-14, MDM4, miRNAs 27a and miR-196a genotyping are depicted in
Table 1.
2.3. Preparation PCR Mix
The PCR reaction was carried out in a total of reaction volume of 12 μL, which was composed of the four primers FO (0.10 μL), RO (0.10 μL), FI (0.10 μL), RI (0.10 μL), (25 pmol of each primer), and 6 μL of green PCR Master Mix (2X) (Cat M712C) (Promega, Madison, WI, USA). The final volume of 12 μL was achieved by using nuclease-free ddH2O. Template DNA (50 ng) was added at the end. The thermocycling procedures were as follows: initial denaturation at 95 °C for 8 min, followed by 32 cycles of denaturation at 95 °C for 33 s; annealing for 35 s at 60 °C for AKT-1 (58 °C), PI3K (59 °C), KLF 14 (56.7 °C) for MDM4 (63 °C) for miRNAs 27a, (61 °C) for miR-196a-2; extension at 72 °C for 40 s, then final extension at 72 °C for 09 min.
2.4. Gel Electrophoresis and PCR Product Visualization
The PCR-amplified products were separated on 2% agarose gel electrophoresis, stained with sybre safe dye, and visualized under UV transilluminator from Bio-Rad, Hercules, CA, USA.
AKT-1 rs1130233 G > A gene variation: The AKT-1 outer region was amplified by the outer primers FO and RO, yielding a band of 466 bp that served as a DNA purity check. A band of 213 bp was produced by the primers FI and RO amplifying the G allele, and a band of 298 bp was produced by the primers FI and RO amplifying the A allele (
Supplementary Figure S1).
Phosphatidylinositol 3-kinase (PI3K) rs121913281 C > T gene variation: The two forward primers and common reverse primer sequence are shown in (
Table 1). Primers F2T and common reverse primers amplify the (TT genotype), that is, the mutant-type allele (364 bp). Primers F1C and the common reverse primer generated a band (364 bp) for the wild type allele (CC genotype) as depicted in
Supplementary Figure S2.
KLF-14 rs972283 G > A gene variation: The KLF14′s outer region was amplified by the outer primers F1 and R1, producing a band of 437 base pairs that serves as a DNA purity control. A band of 221 base pairs (bp) was produced by primers F1 and R2 amplifying the A allele, and a band of 274 bp was produced by primers F2 and R1 amplifying the G allele. Results were confirmed by Sanger sequencing (
Figure 2A–C).
MDM4 rs11801299 A > G gene variation: The exterior primers A band of 468 bp was produced after Fo and R0 amplified the MDM4′s outer region, serving as a DNA purity checker. A band of 223 bp was produced by the A allele by primers FI and Ro, and a band of 301 bp was produced by the G allele by primers Fo and RI (as depicted in
Supplementary Figure S3). The results were confirmed by Sanger sequencing as depicted in
Figure 3A,B.
MiR-27a rs895819 A > G gene variation: The miR-27′s outer region was amplified by the outer primers FO and RO, yielding a band of 353 bp that served as a DNA purity check. A band of 226 bp was produced by primers FI and R2 amplifying the A allele, and a band of 184 bp was produced by primers F2 and R1 amplifying the G allele (as depicted in
Supplementary Figure S4).
MiR-196a2 rs11614913 C > T gene variation: The miR-196a2 exon was flanked by primers FO and RO, which resulted in a 297 bp band that served as a DNA purity check. A band of 153 base pairs (bp) was produced by primers FO and RI amplifying the C allele, and a band of 199 bp was produced by primers FI and RO amplifying the T allele (as depicted in
Supplementary Figure S5).
2.5. Sanger Sequencing for the Confirmation of Genotyping Results
To confirm the genotyping results of KLF-14 rs972283 G > A, MDM4 rs11801299 A > G, PI3K-rs121913281 C > T AKT-1 rs1130233 G > A, and miR-196a2 rs11614913 C > T detected by ARMS-PCR, 20 randomly selected PCR products from the PCR systems for polymorphic sites in these gene were sequenced using Sanger sequencing. Two primers F seq and R Seq were used as sequencing primers as depicted in
Table 1 for the detection of the genotyping in the above genes. The PCR amplification was done followed by purification using QIAquick PCR Purification Kit from Qiagen (Germany). Finally, the purified PCR products were sequenced by Applied Biosystems sequencer.
2.6. Whole Exome Sequencing
For whole exome sequencing, DNA was extracted from peripheral blood using standard Qiagen nucleic acid isolation kits. The library was prepared as per the instruction manual of the Twist 2.0 Exome kit and sequencing was performed using the Illumina NovaSeq 6000 platform as per the user manual. The sequencing reads QC was carried out using FastQC v0.11.9. Raw reads were filtered to remove sequencing adapters and low-quality bases using TrimGalore v0.6.6 software. High quality (HQ) reads thus obtained were mapped on the hg38 human reference genome; variant calling (single nucleotide variants (SNVs), small InDels) was done with the Genome Analysis Toolkit (GATK) v4.2.4.1 software best practice pipeline using haplotype caller.
Variant annotation was carried out using different databases and tools. The RefSeq database was used for identification and characterization of genes associated variants. The disease association for variants was derived using databases like OMIM and ClinVar. The population frequency information from 1000 genomes, ExAC, GnomAD exome, GnomAD genome, and ESP, was used for elimination of common variants/polymorphism. For prediction of implication of coding non-synonymous SNVs on the structure and function of protein, PolyPhen-2 and the SIFT score was used. Furthermore, all variants were separately analyzed by multiple prediction tools for in-silicon variant effect prediction. All variants were then interpreted based on the ACMG guidelines (PMID:25741868) and variants classified as pathogenic, likely pathogenic, and variant of uncertain significance were reported. Some results are presented in the
Supplementary Tables S1–S3.
2.7. Statistical Analysis
All statistical analyses were determined using Med-Calc software, version 20.027 (medcalc.org/calc/odds_ratio.php)/SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) as well as statistical software version 9.4 (SAS Institute, Inc., Cary, NC, USA) and Stata statistical software (StataCorp. 2013. Release 13. College Station, TX, USA). Deflection from Hardy–Weinberg disequilibrium (HWD) was determined by Chi-square (χ2) ‘goodness of fit test’. A p-value < 0.05 is observed as statistically significant difference. The distribution and association of PI3K rs121913281 C > T, AKT-1 rs1130233 G > A, KLF 14 (rs972283 C > T), MDM4 rs11801299 A > G, miRNAs 27a rs895819A > G, and miR-196a-2 rs11614913 C > T alleles and genotypes between the groups was determined by Chi-square test. To assess the association between the risk of BC and the genotypes of KLF 14 (rs972283 C > T), MDM4 (rs11801299 A > G), miRNAs 27a (rs895819A > G), and miR-196a-2 (rs11614913 C > T), we generated odds ratios (ORs), risk ratios (RRs), and risk differences (RDs) with 95% confidence intervals (CIs). The OR was calculated by dividing the probabilities in the first group by the odds in the second group.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}