
Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Renal Clear Cell Carcinoma (ccRCC)
1.2. RCC Statistics
1.3. Risk Factors Associated with ccRCC
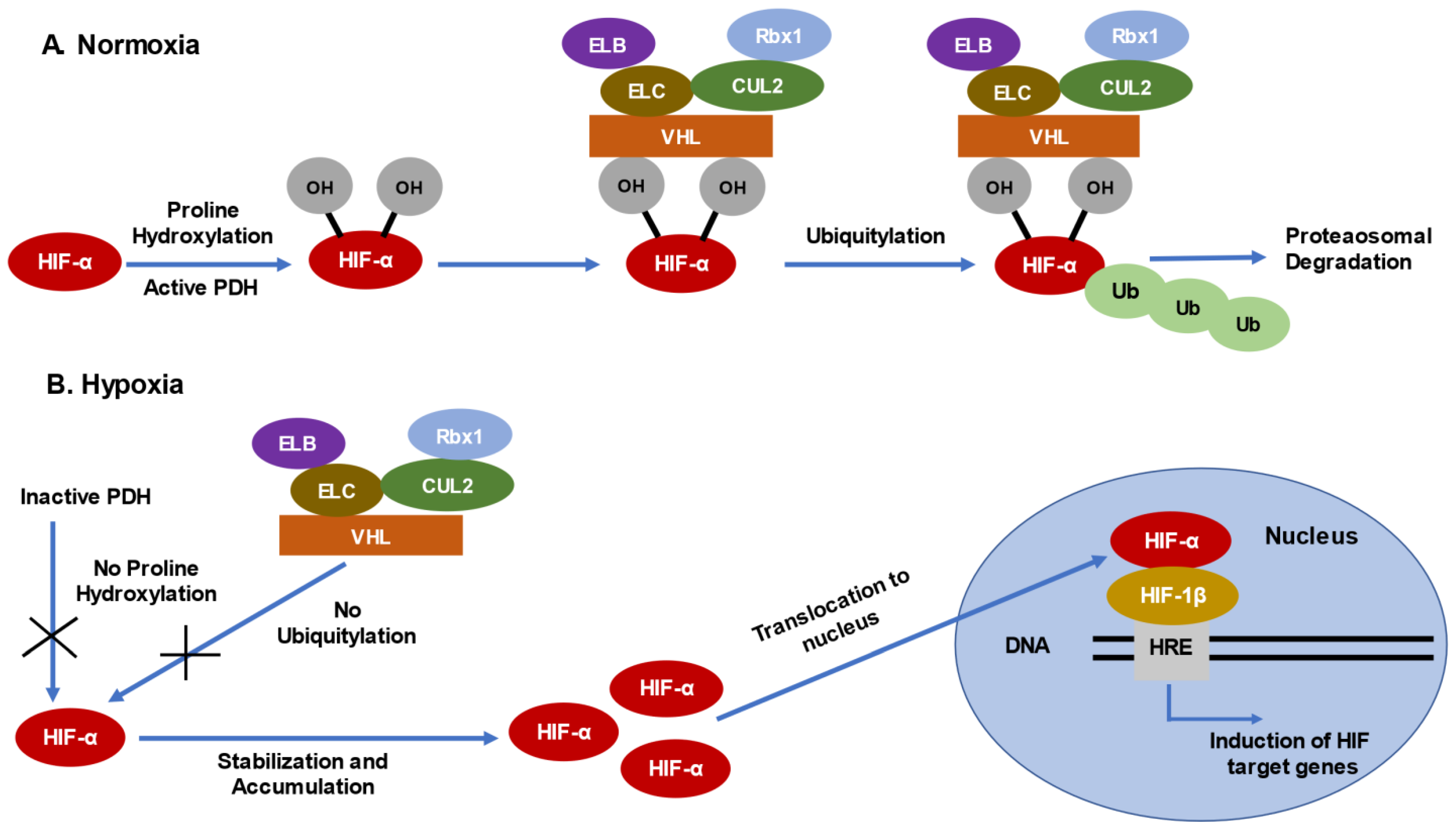
2. Mechanism of Loss of Function of VHL Gene in ccRCC
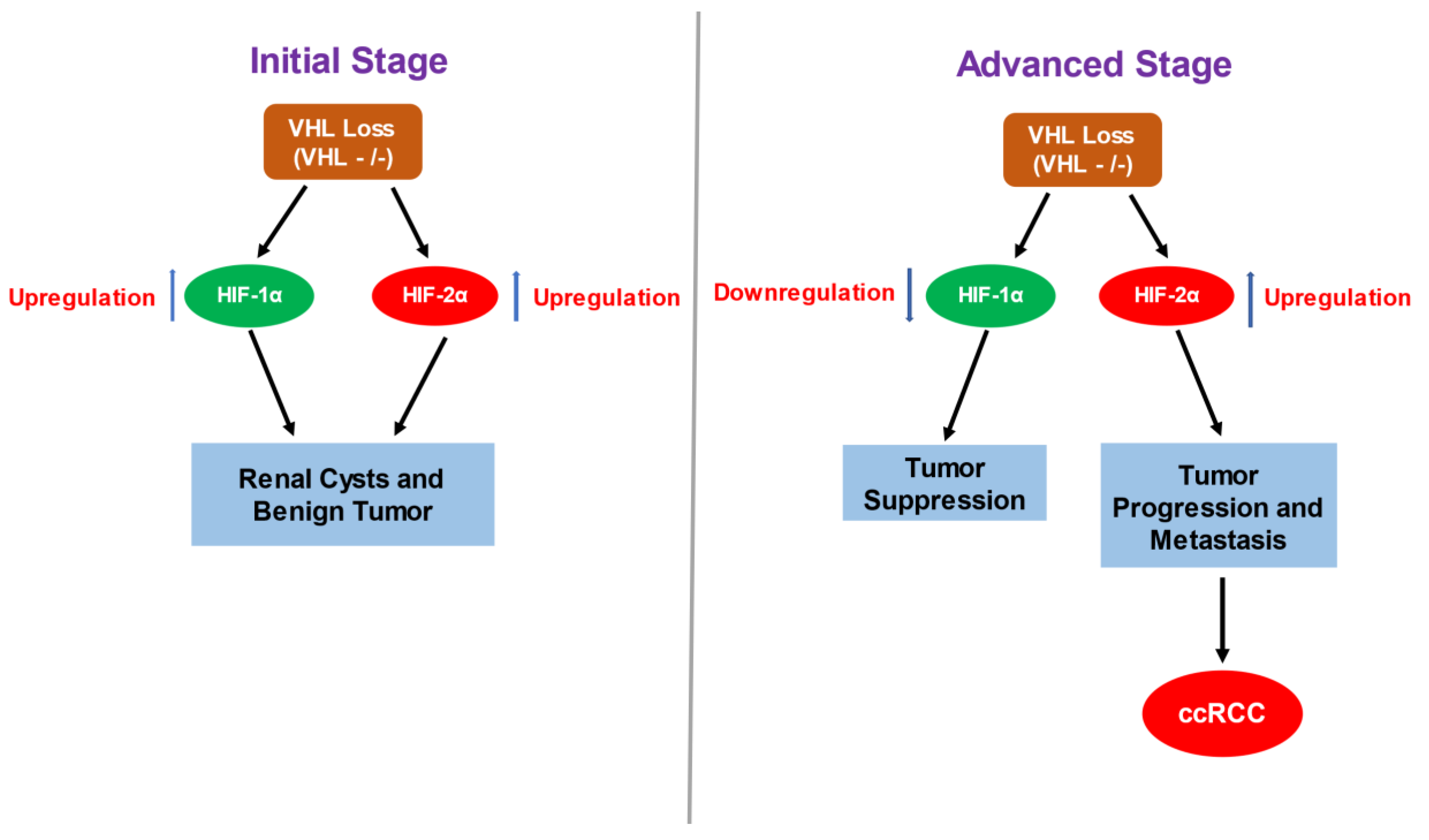
2.1. Consequences of VHL Loss—Stabilization and Accumulation of HIF-α
2.2. HIF-α Involvement in ccRCC—Contrasting Role of HIF-1α and HIF-2α in ccRCC
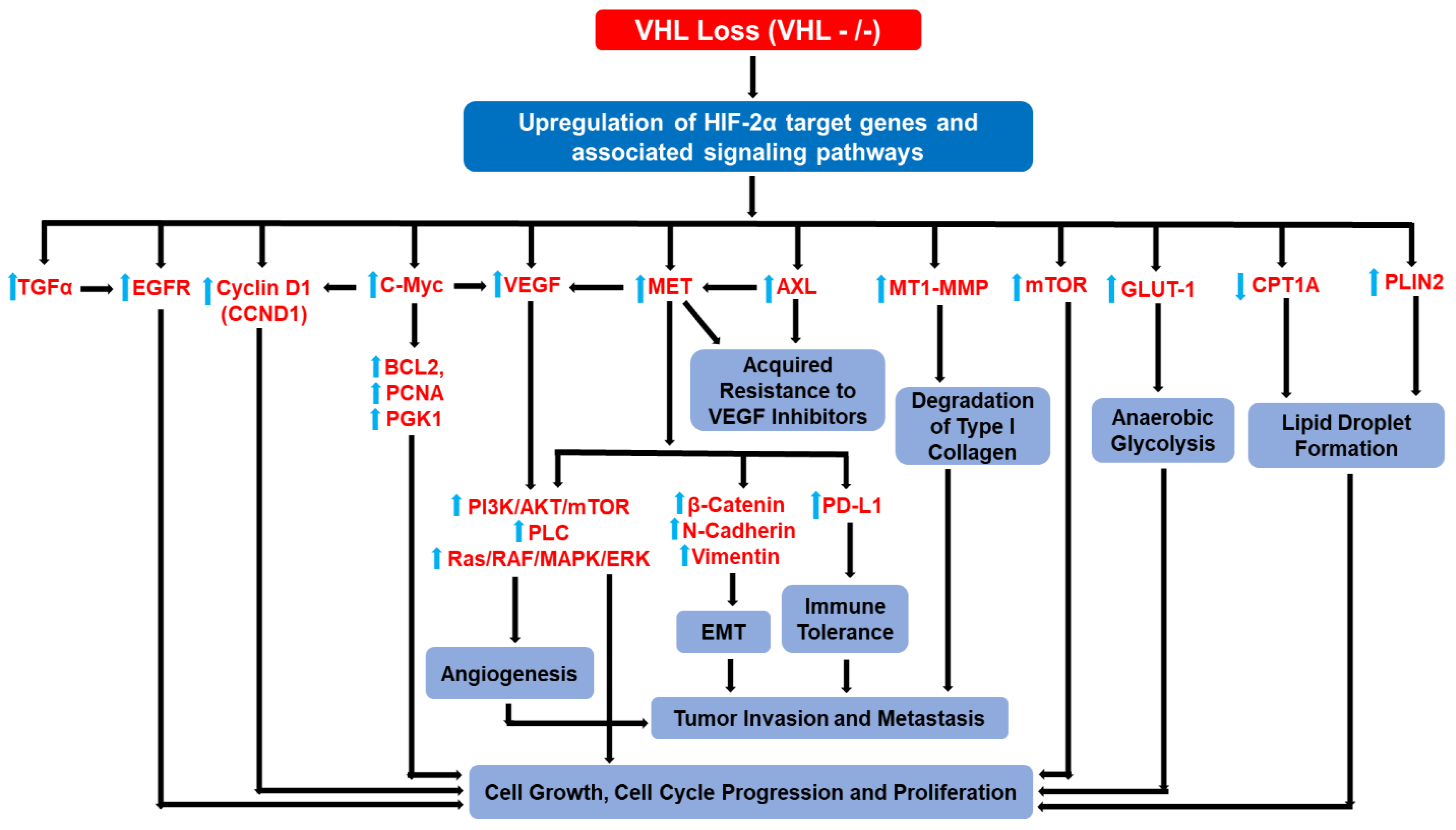
3. HIF-2α Target Genes and HIF-2α-Driven Signaling Pathways Implicated in ccRCC Progression and Metastasis
3.1. TGF-α and EGFR
3.2. c-Myc
3.3. VEGF
3.4. Cyclin D1
3.5. GLUT1
3.6. Carnitine Palmitoyltransferase 1A (CPT1A) and Perilipin 2 (PLIN2)
3.7. MET
3.8. AXL
3.9. MT1-MMP
3.10. PI3K/AKT/mTOR
3.11. Collective Hierarchical Actions of HIF-2α Target Genes
4. Current Treatments, Medical and Scientific Challenges to ccRCC

{kind=link}
{kind=link}
{kind=link}
| Target | Therapy | Citations |
|---|---|---|
| VEGFR | Sorafenib | [113] |
| Sunitinib | [112,182] | |
| Pazopanib | [183] | |
| Cabozantinib | [184] | |
| Axitinib | [185,186] | |
| VEGFR + mTOR | Lenvatinib + Everolimus (anti-mTOR) | [187] |
| VEGF | Bevacizumab + IFN-α (Cytokine) | [188,189] |
| mTOR | Temsirolimus | [190] |
| Everolimus | [191] | |
| PD-1 | Nivolumab | [197,198,199] |
| Pembrolizumab | [200,201,202] | |
| PD-1 + CTLA-4 | Nivolumab + Ipilimumab (anti-CTLA-4) | [210,211,232] |
| PD-1 + VEGFR | Nivolumab + Cabozantinib (anti-VEGFR) | [212,213,214,215] |
| Pembrolizumab + Axitinib (anti-VEGFR) | [216,217,218] | |
| Pembrolizumab + Lenvatinib (anti-VEGFR) | [219] | |
| PD-L1 + VEGFR | Avelumab + Axitinib (anti-VEGFR) | [203,204] |
| HIF-2α | Belzutifan | [192] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cho, H.; Kaelin, W.G. Targeting HIF2 in clear cell renal cell carcinoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avella, C.; Abbosh, P.; Pal, S.K.; Geynisman, D.M. Mutations in renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Frew, I.J.; Moch, H. A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Batavia, A.A.; Schraml, P.; Moch, H. Clear cell renal cell carcinoma with wild-type von Hippel-Lindau gene: A non-existent or new tumour entity? Histopathology 2019, 74, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Ericsson, J.L.; Seljelid, R.; Orrenius, S. Comparative light and electron microscopic observations of the cytoplasmic matrix in renal carcinomas. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1966, 341, 204–223. [Google Scholar] [CrossRef]
- Meléndez-Rodríguez, F.; Roche, O.; Sanchez-Prieto, R.; Aragones, J. Hypoxia-Inducible Factor 2-Dependent Pathways Driving Von Hippel–Lindau-Deficient Renal Cancer. Front. Oncol. 2018, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Makino, T.; Kadomoto, S.; Izumi, K.; Mizokami, A. Epidemiology and Prevention of Renal Cell Carcinoma. Cancers 2022, 14, 4059. [Google Scholar] [CrossRef]
- Bukavina, L.; Bensalah, K.; Bray, F.; Carlo, M.; Challacombe, B.; Karam, J.; Kassouf, W.; Mitchell, T.; Montironi, R.; O’Brien, T. Epidemiology of Renal Cell Carcinoma: 2022 Update. Eur. Urol. 2022, 82, 529–542. [Google Scholar] [CrossRef]
- Atkins, M.B.; Choueiri, T.K. Epidemiology, pathology, and pathogenesis of renal cell carcinoma. UpToDate Retrieved June 2016, 9. [Google Scholar]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019; WHO: Geneva, Switzerland, 2020.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Surveillance Research Program. SEER*Explorer: An Interactive Web-Site for SEER Cancer Statistics; National Cancer Institute: Bethesda, MD, USA, 2021. [Google Scholar]
- American Cancer Society. Cancer Facts & Figures 2023; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- Choyke, P.L.; Glenn, G.M.; Walther, M.; Patronas, N.J.; Linehan, W.M.; Zbar, B. von Hippel-Lindau disease: Genetic, clinical, and imaging features. Radiology 1995, 194, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Maranchie, J.K.; Vasselli, J.R.; Riss, J.; Bonifacino, J.S.; Linehan, W.M.; Klausner, R.D. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002, 1, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Wallen, E.M.; Pruthi, R.S.; Joyce, G.F.; Wise, M.; Project, U.D.i.A. Kidney cancer. J. Urol. 2007, 177, 2006–2019. [Google Scholar] [CrossRef]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.E.; Harris, G.T. Renal cell carcinoma: Diagnosis and management. Am. Fam. Physician 2019, 99, 179–184. [Google Scholar]
- Motzer, R.J.; Bander, N.H.; Nanus, D.M. Renal-cell carcinoma. N. Engl. J. Med. 1996, 335, 865–875. [Google Scholar] [CrossRef]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Speed, J.M.; Trinh, Q.-D.; Choueiri, T.K.; Sun, M. Recurrence in localized renal cell carcinoma: A systematic review of contemporary data. Curr. Urol. Rep. 2017, 18, 15. [Google Scholar] [CrossRef]
- McLaughlin, J.K.; Mandel, J.S.; Blot, W.J.; Schuman, L.M.; Mehl, E.S.; Fraumeni, J.F., Jr. A population-based case-control study of renal cell carcinoma. J. Natl. Cancer Inst. 1984, 72, 275–284. [Google Scholar] [PubMed]
- Yu, M.C.; Mack, T.M.; Hanisch, R.; Cicioni, C.; Henderson, B.E. Cigarette smoking, obesity, diuretic use, and coffee consumption as risk factors for renal cell carcinoma. J. Natl. Cancer Inst. 1986, 77, 351–356. [Google Scholar] [PubMed]
- Ishikawa, I.; Saito, Y.; Asaka, M.; Tomosugi, N.; Yuri, T.; Watanabe, M.; Honda, R. Twenty-year follow-up of acquired renal cystic disease. Clin. Nephrol. 2003, 59, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, L.; Tarone, R.E.; McLaughlin, J.K. The epidemiology of renal cell carcinoma. J. Urol. 2006, 176, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.; Hanbury, D.C.; Kuczyk, M.A.; Merseburger, A.S.; Mulders, P.F.; Patard, J.-J.; Sinescu, I.C. Renal cell carcinoma guideline. Eur. Urol. 2007, 51, 1502–1510. [Google Scholar] [CrossRef]
- Rakowski, S.; Winterkorn, E.; Paul, E.; Steele, D.; Halpern, E.F.; Thiele, E. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.-M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Gnarra, J.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.-M. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Zbar, B.; Brauch, H.; Talmadge, C.; Linehan, M. Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 1987, 327, 721–724. [Google Scholar] [CrossRef]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Hakimi, A.A.; Pham, C.G.; Hsieh, J.J. A clear picture of renal cell carcinoma. Nat. Genet. 2013, 45, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Kaelin Jr, W.G.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Lisztwan, J.; Imbert, G.; Wirbelauer, C.; Gstaiger, M.; Krek, W. The von Hippel–Lindau tumor suppressor protein is a component of an E3 ubiquitin–protein ligase activity. Genes Dev. 1999, 13, 1822–1833. [Google Scholar] [CrossRef]
- Duan, D.R.; Pause, A.; Burgess, W.H.; Aso, T.; Chen, D.Y.; Garrett, K.P.; Conaway, R.C.; Conaway, J.W.; Linehan, W.M.; Klausner, R.D. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science 1995, 269, 1402–1406. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kaelin Jr, W.G. The von Hippel–Lindau tumor suppressor protein. Curr. Opin. Genet. Dev. 2001, 11, 27–34. [Google Scholar] [CrossRef]
- Lonergan, K.M.; Iliopoulos, O.; Ohh, M.; Kamura, T.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol. 1998, 18, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Pause, A.; Lee, S.; Worrell, R.A.; Chen, D.Y.; Burgess, W.H.; Linehan, W.M.; Klausner, R.D. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 2156–2161. [Google Scholar] [CrossRef] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 617–626. [Google Scholar] [CrossRef]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.; Dachs, G.; Gleadle, J.; Nicholls, L.; Harris, A.; Stratford, I.; Hankinson, O.; Pugh, C.; Ratcliffe, P. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-S.; Vortmeyer, A.O.; Lubensky, I.A.; Vogel, T.W.; Ikejiri, B.; Ferlicot, S.; Benoît, G.; Giraud, S.; Oldfield, E.H.; Linehan, W.M. Coexpression of erythropoietin and erythropoietin receptor in von hippel-lindau disease–associated renal cysts and renal cell carcinoma. Clin. Cancer Res. 2005, 11, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.; Pham, T.; Hanby, A.; Frantz, G.; Peale, F.; Wu, T.; Koeppen, H.; Hillan, K. Expression of vascular endothelial growth factor, hypoxia inducible factor 1α, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 2004, 57, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef] [Green Version]
- Gnarra, J.R.; Zhou, S.; Merrill, M.J.; Wagner, J.R.; Krumm, A.; Papavassiliou, E.; Oldfield, E.H.; Klausner, R.D.; Linehan, W.M. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 10589–10594. [Google Scholar] [CrossRef] [Green Version]
- Siemeister, G.; Weindel, K.; Mohrs, K.; Barleon, B.; Martiny-Baron, G.; Marmé, D. Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Res. 1996, 56, 2299–2301. [Google Scholar]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin Jr, W.G. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönenberger, D.; Harlander, S.; Rajski, M.; Jacobs, R.A.; Lundby, A.-K.; Adlesic, M.; Hejhal, T.; Wild, P.J.; Lundby, C.; Frew, I.J. Formation of renal cysts and tumors in Vhl/Trp53-deficient mice requires HIF1α and HIF2α. Cancer Res. 2016, 76, 2025–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandriota, S.J.; Turner, K.J.; Davies, D.R.; Murray, P.G.; Morgan, N.V.; Sowter, H.M.; Wykoff, C.C.; Maher, E.R.; Harris, A.L.; Ratcliffe, P.J. HIF activation identifies early lesions in VHL kidneys: Evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002, 1, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Rosenberger, C.; Mandriota, S.; Jürgensen, J.S.; Wiesener, M.S.; Hörstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.-U. Expression of hypoxia-inducible factor-1α and-2α in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdue, M.P.; Johansson, M.; Zelenika, D.; Toro, J.R.; Scelo, G.; Moore, L.E.; Prokhortchouk, E.; Wu, X.; Kiemeney, L.A.; Gaborieau, V. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat. Genet. 2011, 43, 60–65. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Kawanami, D.; Mahabeleshwar, G.H.; Lin, Z.; Atkins, G.B.; Hamik, A.; Haldar, S.M.; Maemura, K.; LaManna, J.C.; Jain, M.K. Kruppel-like factor 2 inhibits hypoxia-inducible factor 1α expression and function in the endothelium. J. Biol. Chem. 2009, 284, 20522–20530. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP Selectively Mediate Ubiquitination and Degradation of Hypoxia-inducible Factor (HIF)-1α but Not HIF-2α 2. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef]
- Koh, M.Y.; Darnay, B.G.; Powis, G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1α, leading to its oxygen-independent degradation. Mol. Cell. Biol. 2008, 28, 7081–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Lemos Jr, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α-to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Nguyen, V.; Lemos, R.; Darnay, B.G.; Kiriakova, G.; Abdelmelek, M.; Ho, T.H.; Karam, J.; Monzon, F.A.; Jonasch, E. Hypoxia-Induced SUMOylation of E3 Ligase HAF Determines Specific Activation of HIF2 in Clear-Cell Renal Cell CarcinomaHAF SUMOylation Drives HIF2 in CRCC. Cancer Res. 2015, 75, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Wu, Y.; Jiang, W. MicroRNA-18a decreases choroidal endothelial cell proliferation and migration by inhibiting HIF1A expression. Med. Sci. Monit. 2015, 21, 1642–1647. [Google Scholar]
- Jaśkiewicz, M.; Moszyńska, A.; Króliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzyńska, A.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell. Mol. Biol. Lett. 2022, 27, 109. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin Jr, W.G.; Kemp, C. Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, e83. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, M.; Doucette, D.; Siddiqui, N.; Iliopoulos, O. Inhibition of Hypoxia-Inducible Factor Is Sufficient for Growth Suppression of VHL−/− Tumors. Mol. Cancer Res. 2004, 2, 89–95. [Google Scholar] [CrossRef]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Đorđević, G.; Matušan Ilijaš, K.; Hadžisejdić, I.; Maričić, A.; Grahovac, B.; Jonjić, N. EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J. Biomed. Sci. 2012, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Thomasson, M.; Hedman, H.; Ljungberg, B.; Henriksson, R. Gene expression pattern of the epidermal growth factor receptor family and LRIG1 in renal cell carcinoma. BMC Res. Notes 2012, 5, 216. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Roche, O.; Yan, M.S.; Finak, G.; Evans, A.J.; Metcalf, J.L.; Hast, B.E.; Hanna, S.C.; Wondergem, B.; Furge, K.A. Regulation of endocytosis via the oxygen-sensing pathway. Nat. Med. 2009, 15, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, H. The von Hippel-Lindau tumor suppressor protein promotes c-Cbl-independent poly-ubiquitylation and degradation of the activated EGFR. PLoS ONE 2011, 6, e23936. [Google Scholar] [CrossRef] [PubMed]
- Ramp, U.; Jaquet, K.; Reinecke, P.; Schardt, C.; Friebe, U.; Nitsch, T.; Marx, N.; Gabbert, H.E.; Gerharz, C.-D. Functional intactness of stimulatory and inhibitory autocrine loops in human renal carcinoma cell lines of the clear cell type. J. Urol. 1997, 157, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Knebelmann, B.; Ananth, S.; Cohen, H.T.; Sukhatme, V.P. Transforming growth factor α is a target for the von Hippel-Lindau tumor suppressor. Cancer Res. 1998, 58, 226–231. [Google Scholar] [PubMed]
- De Paulsen, N.; Brychzy, A.; Fournier, M.-C.; Klausner, R.D.; Gnarra, J.R.; Pause, A.; Lee, S. Role of transforming growth factor-α in von Hippel–Lindau (VHL)−/− clear cell renal carcinoma cell proliferation: A possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1387–1392. [Google Scholar] [PubMed]
- Gunaratnam, L.; Morley, M.; Franovic, A.; De Paulsen, N.; Mekhail, K.; Parolin, D.A.; Nakamura, E.; Lorimer, I.A.; Lee, S. Hypoxia inducible factor activates the transforming growth factor-α/epidermal growth factor receptor growth stimulatory pathway in VHL−/− renal cell carcinoma cells. J. Biol. Chem. 2003, 278, 44966–44974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.; Gunaratnam, L.; Morley, M.; Franovic, A.; Mekhail, K.; Lee, S. Silencing of Epidermal Growth Factor Receptor Suppresses Hypoxia-Inducible Factor-2–Driven VHL−/− Renal Cancer. Cancer Res. 2005, 65, 5221–5230. [Google Scholar] [CrossRef] [Green Version]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [Green Version]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-W.; Chang, W.-H.; Su, Y.-C.; Chen, Y.-C.; Lai, Y.-H.; Wu, P.-T.; Hsu, C.-I.; Lin, W.-C.; Lai, M.-K.; Lin, J.-Y. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009, 273, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.M.; Malek, R.L.; Kim, S.; Chiao, C.; He, M.; Ruffy, M.; Sanka, K.; Lee, N.H.; Dang, C.V.; Liu, E.T. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000, 60, 5922–5928. [Google Scholar] [PubMed]
- Zhou, Q.; Hopp, T.; Fuqua, S.; Steeg, P.S. Cyclin D1 in breast premalignancy and early breast cancer: Implications for prevention and treatment. Cancer Lett. 2001, 162, 3–17. [Google Scholar] [CrossRef]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Kato, T.; Kameoka, S.; Kimura, T.; Nishikawa, T.; Kobayashi, M. C-erbB-2 and PCNA as prognostic indicators of long-term survival in breast cancer. Anticancer. Res. 2002, 22, 1097–1103. [Google Scholar]
- Lay, A.J.; Jiang, X.-M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 2000, 408, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Veikkola, T.; Karkkainen, M.; Claesson-Welsh, L.; Alitalo, K. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 2000, 60, 203–212. [Google Scholar] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B. HIF-α effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Fong, G.H. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis 2008, 11, 121–140. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Sosman, J.A.; Motzer, R.J. Therapy targeted at vascular endothelial growth factor in metastatic renal cell carcinoma: Biology, clinical results and future development. BJU Int. 2005, 96, 286–290. [Google Scholar] [CrossRef]
- Jacobsen, J.; Grankvist, K.; Rasmuson, T.; Bergh, A.; Landberg, G.; Ljungberg, B. Expression of vascular endothelial growth factor protein in human renal cell carcinoma. BJU Int. 2004, 93, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Minardi, D.; Santoni, M.; Lucarini, G.; Mazzucchelli, R.; Burattini, L.; Conti, A.; Bianconi, M.; Scartozzi, M.; Milanese, G.; Di Primio, R. Tumor VEGF expression correlates with tumor stage and identifies prognostically different groups in patients with clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 113.e111–113.e117. [Google Scholar] [CrossRef] [PubMed]
- Baldewijns, M.; Thijssen, V.; Van den Eynden, G.; Van Laere, S.; Bluekens, A.; Roskams, T.; Van Poppel, H.; De Bruine, A.; Griffioen, A.; Vermeulen, P. High-grade clear cell renal cell carcinoma has a higher angiogenic activity than low-grade renal cell carcinoma based on histomorphological quantification and qRT–PCR mRNA expression profile. Br. J. Cancer 2007, 96, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Huang, J.; Chen, J.; Wang, R.; Dong, C.; Lu, S.; Wu, X. Expression and significance of FOXP1, HIF-1a and VEGF in renal clear cell carcinoma. J. Buon 2015, 20, 188–195. [Google Scholar]
- Zhang, T.; Niu, X.; Liao, L.; Cho, E.-A.; Yang, H. The contributions of HIF-target genes to tumor growth in RCC. PLoS ONE 2013, 8, e80544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Shinojima, T.; Oya, M.; Takayanagi, A.; Mizuno, R.; Shimizu, N.; Murai, M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1α expression maintain vascular endothelial growth factor expression through HIF-2α. Carcinogenesis 2007, 28, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti–vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Lai, Y.; Zhao, Z.; Zeng, T.; Liang, X.; Chen, D.; Duan, X.; Zeng, G.; Wu, W. Crosstalk between VEGFR and other receptor tyrosine kinases for TKI therapy of metastatic renal cell carcinoma. Cancer Cell Int. 2018, 18, 31. [Google Scholar] [CrossRef]
- Barnes, D.M.; Gillett, C.E. Cyclin D1 in breast cancer. Breast Cancer Res. Treat. 1998, 52, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Simpson, D.J.; Frost, S.J.; Bicknell, J.E.; Broome, J.C.; McNicol, A.M.; Clayton, R.N.; Farrell, W.E. Aberrant expression of G 1/S regulators is a frequent event in sporadic pituitary adenomas. Carcinogenesis 2001, 22, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeguchi, M.; Sakatani, T.; Ueta, T.; Kaibara, N. Cyclin D1 expression and retinoblastoma gene protein (pRB) expression in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2001, 127, 531–536. [Google Scholar] [CrossRef]
- Gansauge, S.; Gansauge, F.; Ramadani, M.; Stobbe, H.; Rau, B.; Harada, N.; Beger, H.G. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997, 57, 1634–1637. [Google Scholar]
- Yamanouchi, H.; Furihata, M.; Fujita, J.; Murakami, H.; Yoshinouchi, T.; Takahara, J.; Ohtsuki, Y. Expression of cyclin E and cyclin D1 in non-small cell lung cancers. Lung Cancer 2001, 31, 3–8. [Google Scholar] [CrossRef]
- Donnellan, R.; Chetty, R. Cyclin D1 and human neoplasia. Mol. Pathol. 1998, 51, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jiping, Z.; Like, Y.; Ping, Z.; Yong, S.; Qin, W. The relationships between cyclin D1 expression and prognosis of non-small cell lung cancer. Chin. J. Lung Cancer 2010, 13, 803–808. [Google Scholar]
- Hedberg, Y.; Ljungberg, B.; Roos, G.; Landberg, G. Expression of cyclin D1, D3, E, and p27 in human renal cell carcinoma analysed by tissue microarray. Br. J. Cancer 2003, 88, 1417–1423. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Vasselli, J.R.; Stearman, R.; Linehan, W.M.; Klausner, R.D. VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res. 2002, 62, 3014–3019. [Google Scholar] [PubMed]
- Zatyka, M.; da Silva, N.F.; Clifford, S.C.; Morris, M.R.; Wiesener, M.S.; Eckardt, K.-U.; Houlston, R.S.; Richards, F.M.; Latif, F.; Maher, E.R. Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease. Cancer Res. 2002, 62, 3803–3811. [Google Scholar] [PubMed]
- Lin, B.; Brynes, R.K.; Gelb, A.B.; McCourty, A.; Amin, M.B.; Medeiros, L.J. Cyclin D1 expression in renal carcinomas and oncocytomas: An immunohistochemical study. Mod. Pathol. 1998, 11, 1075–1081. [Google Scholar]
- Wang, Q.s.; Li, F.; Liao, Z.q.; Li, K.; Yang, X.l.; Lin, Y.y.; Zhao, Y.l.; Weng, S.y.; Xia, Y.; Ye, Y. Low level of Cyclin-D1 correlates with worse prognosis of clear cell renal cell carcinoma patients. Cancer Med. 2019, 8, 4100–4109. [Google Scholar] [CrossRef]
- Kotulak-Chrzaszcz, A.; Rybarczyk, A.; Klacz, J.; Matuszewski, M.; Kmiec, Z.; Wierzbicki, P.M. Expression levels of sonic hedgehog pathway genes and their targets are upregulated in early clear cell renal cell carcinoma. Int. J. Mol. Med. 2022, 49, 58. [Google Scholar] [CrossRef]
- Rizwan, M.; Farooq, N.; Tariq, H.; Akhtar, A.N.; Ibne-Rasa, S.N.; Naz, S. To Study the Association of Cyclin D1 Expression with Fuhrman’s Nuclear Grading in Renal Cell Carcinoma. Pak. J. Med. Health Sci. 2022, 16, 96. [Google Scholar] [CrossRef]
- Schödel, J.; Bardella, C.; Sciesielski, L.K.; Brown, J.M.; Pugh, C.W.; Buckle, V.; Tomlinson, I.P.; Ratcliffe, P.J.; Mole, D.R. Common genetic variants at the 11q13. 3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet. 2012, 44, 420–425. [Google Scholar] [CrossRef]
- Chen, D.; Sun, X.; Zhang, X.; Cao, J. Inhibition of the CDK4/6-cyclin D-Rb pathway by ribociclib augments chemotherapy and immunotherapy in renal cell carcinoma. BioMed Res. Int. 2020, 2020, 9525207. [Google Scholar] [CrossRef]
- Karim, S.; Al-Maghrabi, J.A.; Farsi, H.M.; Al-Sayyad, A.J.; Schulten, H.-J.; Buhmeida, A.; Mirza, Z.; Al-Boogmi, A.A.; Ashgan, F.T.; Shabaad, M.M. Cyclin D1 as a therapeutic target of renal cell carcinoma-a combined transcriptomics, tissue microarray and molecular docking study from the Kingdom of Saudi Arabia. BMC Cancer 2016, 16, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragonés, J.; Fraisl, P.; Baes, M.; Carmeliet, P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009, 9, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, S.-Y.; Hao, Y.-B.; Nan, K.-J.; Fan, T.-L. Cancer stem cells niche: A target for novel cancer therapeutics. Cancer Treat. Rev. 2013, 39, 290–296. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [Green Version]
- Ambrosetti, D.; Dufies, M.; Dadone, B.; Durand, M.; Borchiellini, D.; Amiel, J.; Pouyssegur, J.; Rioux-Leclercq, N.; Pages, G.; Burel-Vandenbos, F. The two glycolytic markers GLUT1 and MCT1 correlate with tumor grade and survival in clear-cell renal cell carcinoma. PLoS ONE 2018, 13, e0193477. [Google Scholar] [CrossRef] [Green Version]
- Maranchie, J.K.; Zhan, Y. Nox4 is critical for hypoxia-inducible factor 2-α transcriptional activity in von Hippel-Lindau–deficient renal cell carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef] [Green Version]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Büttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlösser, H.; Hartmann, A. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8+ T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef]
- Yamasaki, T.; Seki, N.; Yoshino, H.; Itesako, T.; Yamada, Y.; Tatarano, S.; Hidaka, H.; Yonezawa, T.; Nakagawa, M.; Enokida, H. Tumor-suppressive micro RNA-1291 directly regulates glucose transporter 1 in renal cell carcinoma. Cancer Sci. 2013, 104, 1411–1419. [Google Scholar] [CrossRef]
- Liu, M.; Gao, J.; Huang, Q.; Jin, Y.; Wei, Z. Downregulating microRNA-144 mediates a metabolic shift in lung cancer cells by regulating GLUT1 expression. Oncol. Lett. 2016, 11, 3772–3776. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Hu, J.-W.; Xiong, W.-J.; Mi, J. miR-186 regulates glycolysis through Glut1 during the formation of cancer-associated fibroblasts. Asian Pac. J. Cancer Prev. 2014, 15, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.; Dias, F.; Nogueira, I.; Leão, A.; Gonçalves, N.; Araújo, L.; Granja, S.; Baltazar, F.; Teixeira, A.L.; Medeiros, R. Cancer cells’ metabolism dynamics in renal cell carcinoma patients’ outcome: Influence of GLUT-1-Related hsa-miR-144 and hsa-miR-186. Cancers 2021, 13, 1733. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [Green Version]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Pérez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; López-Larrubia, P. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Pisters, L.L.; Troncoso, P.; Zhau, H.E.; Li, W.; von Eschenbach, A.C.; Chung, L.W. c-Met proto-oncogene expression in benign and malignant human prostate tissues. J. Urol. 1995, 154, 293–298. [Google Scholar] [CrossRef]
- Natali, P.G.; Prat, M.; Nicotra, M.R.; Bigotti, A.; Olivero, M.; Comoglio, P.M.; Di Renzo, M.F. Overexpression of the met/HGF receptor in renal cell carcinomas. Int. J. Cancer 1996, 69, 212–217. [Google Scholar] [CrossRef]
- HORIE, S.; ARUGA, S.; KAWAMATA, H.; OKUI, N.; KAKIZOE, T.; KITAMURA, T. Biological role of HGF/MET pathway in renal cell carcinoma. J. Urol. 1999, 161, 990–997. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.-L.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Longati, P.; Gramaglia, D.; Stella, M.C.; Comoglio, P.M. Gab1 coupling to the HGF/Met receptor multifunctional docking site requires binding of Grb2 and correlates with the transforming potential. Oncogene 1997, 15, 3103–3111. [Google Scholar] [CrossRef] [Green Version]
- Behbahani, T.E.; Thierse, C.; Baumann, C.; Holl, D.; Bastian, P.J.; von Ruecker, A.; Müller, S.C.; Ellinger, J.; Hauser, S. Tyrosine kinase expression profile in clear cell renal cell carcinoma. World J. Urol. 2012, 30, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gordoa, T.; García-Bermejo, M.L.; Grande, E.; Garrido, P.; Carrato, A.; Molina-Cerrillo, J. Targeting tyrosine kinases in renal cell carcinoma:“new bullets against old guys”. Int. J. Mol. Sci. 2019, 20, 1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, A.; Kubota, T.; Taiyoh, H.; Fujiwara, H.; Okamoto, K.; Ichikawa, D.; Shiozaki, A.; Komatsu, S.; Nakanishi, M.; Kuriu, Y. HGF regulates VEGF expression via the c-Met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int. J. Oncol. 2013, 42, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaigawa, N.; Yao, M.; Baba, M.; Kato, S.; Kishida, T.; Hattori, K.; Nagashima, Y.; Kubota, Y. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006, 66, 3699–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koochekpour, S.; Jeffers, M.; Wang, P.H.; Gong, C.; Taylor, G.A.; Roessler, L.M.; Stearman, R.; Vasselli, J.R.; Stetler-Stevenson, W.G.; Kaelin Jr, W.G. The von Hippel-Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol. Cell. Biol. 1999, 19, 5902–5912. [Google Scholar] [CrossRef] [Green Version]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.-D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Shin, S.-J.; Jeon, Y.K.; Kim, P.-J.; Cho, Y.M.; Koh, J.; Chung, D.H.; Go, H. Clinicopathologic analysis of PD-L1 and PD-L2 expression in renal cell carcinoma: Association with oncogenic proteins status. Ann. Surg. Oncol. 2016, 23, 694–702. [Google Scholar] [CrossRef]
- Kammerer-Jacquet, S.-F.; Medane, S.; Bensalah, K.; Bernhard, J.-C.; Yacoub, M.; Dupuis, F.; Ravaud, A.; Verhoest, G.; Mathieu, R.; Peyronnet, B. Correlation of c-MET expression with PD-L1 expression in metastatic clear cell renal cell carcinoma treated by sunitinib first-line therapy. Target. Oncol. 2017, 12, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Balan, M.; y Teran, E.M.; Waaga-Gasser, A.M.; Gasser, M.; Choueiri, T.K.; Freeman, G.; Pal, S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J. Biol. Chem. 2015, 290, 8110–8120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Chen, L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014, 20, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C.E. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel–Lindau renal cell carcinoma. Oncogene 2005, 24, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Seiki, M. Membrane-type 1 matrix metalloproteinase: A key enzyme for tumor invasion. Cancer Lett. 2003, 194, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Seiki, M.; Yana, I. Roles of pericellular proteolysis by membrane type-1 matrix metalloproteinase in cancer invasion and angiogenesis. Cancer Sci. 2003, 94, 569–574. [Google Scholar] [CrossRef]
- Sounni, N.E.; Noël, A. Membrane type-matrix metalloproteinases and tumor progression. Biochimie 2005, 87, 329–342. [Google Scholar] [CrossRef]
- Petrella, B.L.; Brinckerhoff, C.E. Tumor cell invasion of von Hippel Lindau renal cell carcinoma cells is mediated by membrane type-1 matrix metalloproteinase. Mol. Cancer 2006, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Ganner, A.; Gehrke, C.; Klein, M.; Thegtmeier, L.; Matulenski, T.; Wingendorf, L.; Wang, L.; Pilz, F.; Greidl, L.; Meid, L. VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma. Sci. Rep. 2021, 11, 14827. [Google Scholar] [CrossRef]
- Pantuck, A.J.; Seligson, D.B.; Klatte, T.; Yu, H.; Leppert, J.T.; Moore, L.; O’Toole, T.; Gibbons, J.; Belldegrun, A.S.; Figlin, R.A. Prognostic relevance of the mTOR pathway in renal cell carcinoma: Implications for molecular patient selection for targeted therapy. Cancer 2007, 109, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Doan, H.; Parsons, A.; Devkumar, S.; Selvarajah, J.; Miralles, F.; Carroll, V.A. Hif-Mediated suppression of DEPTOR confers resistance to mTOR kinase inhibition in renal cancer. Iscience 2019, 21, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Bhayani, S.; Bro, W.P.; Chang, S.S.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; Fishman, M. Kidney cancer, version 2.2017, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 804–834. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Rini, B.I.; Beckermann, K.E. Emerging Targets in Clear Cell Renal Cell Carcinoma. Cancers 2022, 14, 4843. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posadas, E.M.; Limvorasak, S.; Figlin, R.A. Targeted therapies for renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 496–511. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.-L.; Peltola, K. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Ou, S.-S.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J. Clin. Oncol. 2008, 26, 5422–5428. [Google Scholar] [CrossRef]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P.; Dutcher, J. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: Final results of CALGB 90206. J. Clin. Oncol. 2010, 28, 2137–2143. [Google Scholar] [CrossRef] [Green Version]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Fallah, J.; Brave, M.H.; Weinstock, C.; Mehta, G.U.; Bradford, D.; Gittleman, H.; Bloomquist, E.W.; Charlab, R.; Hamed, S.S.; Miller, C.P. FDA Approval Summary: Belzutifan for von Hippel-Lindau Disease–Associated Tumors. Clin. Cancer Res. 2022, 28, 4843–4848. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Rumble, R.B.; Van Veldhuizen, P.J.; Al-Ahmadie, H.; Emamekhoo, H.; Hauke, R.J.; Louie, A.V.; Milowsky, M.I.; Molina, A.M.; Rose, T.L. Management of metastatic clear cell renal cell carcinoma: ASCO guideline. J. Clin. Oncol. 2022, 40, 2957–2995. [Google Scholar] [CrossRef]
- Tenold, M.; Ravi, P.; Kumar, M.; Bowman, A.; Hammers, H.; Choueiri, T.K.; Lara, P.N., Jr. Current approaches to the treatment of advanced or metastatic renal cell carcinoma. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2020; Volume 40, pp. 187–196. [Google Scholar]
- Jahangir, M.; Yazdani, O.; Kahrizi, M.S.; Soltanzadeh, S.; Javididashtbayaz, H.; Mivefroshan, A.; Ilkhani, S.; Esbati, R. Clinical potential of PD-1/PD-L1 blockade therapy for renal cell carcinoma (RCC): A rapidly evolving strategy. Cancer Cell Int. 2022, 22, 401. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Escudier, B.; Sharma, P.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. CheckMate 025 randomized phase 3 study: Outcomes by key baseline factors and prior therapy for nivolumab versus everolimus in advanced renal cell carcinoma. Eur. Urol. 2017, 72, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Motzer, R.J.; Sharma, P.; Wagstaff, J.; Plimack, E.R.; Hammers, H.J.; Donskov, F.; Gurney, H.; Sosman, J.A.; Zalewski, P.G. Treatment beyond progression in patients with advanced renal cell carcinoma treated with nivolumab in CheckMate 025. Eur. Urol. 2017, 72, 368–376. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Quinn, D.I.; Zhang, T.; Gurney, H.; Doshi, G.K.; Cobb, P.W.; Parnis, F.; Lee, J.-L.; Park, S.H.; Semenov, A. KEYNOTE-564: A phase 3, randomized, double blind, trial of pembrolizumab in the adjuvant treatment of renal cell carcinoma. Am. Soc. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Powles, T.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Symeonides, S.N.; Hajek, J.; Gurney, H.; Chang, Y.-H.; Lee, J.L. Pembrolizumab versus placebo as post-nephrectomy adjuvant therapy for clear cell renal cell carcinoma (KEYNOTE-564): 30-month follow-up analysis of a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022, 23, 1133–1144. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.-H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N. Adjuvant pembrolizumab after nephrectomy in renal-cell carcinoma. N. Engl. J. Med. 2021, 385, 683–694. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic MelanomaIpilimumab for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 2007, 30, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albiges, L.; Powles, T.; Staehler, M.; Bensalah, K.; Giles, R.H.; Hora, M.; Kuczyk, M.A.; Lam, T.B.; Ljungberg, B.; Marconi, L.; et al. Updated European Association of Urology guidelines on renal cell carcinoma: Immune checkpoint inhibition is the new backbone in first-line treatment of metastatic clear-cell renal cell carcinoma. Eur. Urol. 2019, 76, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Hammers, H.J.; Plimack, E.R.; Infante, J.R.; Rini, B.I.; McDermott, D.F.; Lewis, L.D.; Voss, M.H.; Sharma, P.; Pal, S.K.; Razak, A.R.; et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: The CheckMate 016 study. J. Clin. Oncol. 2017, 35, 3851–3858. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Apolo, A.B.; Powles, T.; Escudier, B.; Aren, O.R.; Shah, A.; Kessler, E.R.; Hsieh, J.J.; Zhang, J.; Simsek, B.; et al. A phase 3, randomized, open-label study of nivolumab combined with cabozantinib vs sunitinib in patients with previously untreated advanced or metastatic renal cell carcinoma (RCC.; CheckMate 9ER). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Kfoury, M.; Oing, C. ESMO20 YO for YO: Highlights on metastatic renal cell carcinoma—The CheckMate-9ER trial. ESMO Open 2021, 6, 100025. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. New Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Khalil, N.; Sarkis, J.; Abi Tayeh, G. Use of immunotherapy with programmed cell death 1 vs programmed cell death ligand 1 in renal cell carcinoma: Lessons from CheckMate 9ER and IMmotion 151. J. Oncol. Pharm. Pract. 2021, 27, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Bilusic, M. Pembrolizumab in combination with axitinib as first-line treatment for patients with renal cell carcinoma (RCC): Evidence to date. Cancer Manag. Res. 2020, 12, 7321–7330. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Guillaume, Z.; Auvray, M.; Vano, Y.; Oudard, S.; Helley, D.; Mauge, L. Renal Carcinoma and Angiogenesis: Therapeutic Target and Biomarkers of Response in Current Therapies. Cancers 2022, 14, 6167. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lu, T.; Lu, S.; Ma, S.; Han, D.; Zhang, K.; Xu, C.; Liu, S.; Gan, L.; Wu, X.; et al. Single-cell analysis of multiple cancer types reveals differences in endothelial cells between tumors and normal tissues. Comput. Struct. Biotechnol. J. 2022, 21, 665–676. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Dudani, S.; Savard, M.-F.; Heng, D.Y. An update on predictive biomarkers in metastatic renal cell carcinoma. Eur. Urol. Focus 2020, 6, 34–36. [Google Scholar] [CrossRef]
- Baldewijns, M.M.; van Vlodrop, I.J.; Vermeulen, P.B.; Soetekouw, P.M.; van Engeland, M.; de Bruïne, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Philips, G.K.; Atkins, M.B. New agents and new targets for renal cell carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e222–e227. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.A.; Gupta, G.N.; Srinivasan, R. Update on targeted therapies for clear cell renal cell carcinoma. Curr. Opin. Oncol. 2011, 23, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, A.; Chisholm, G. The natural history of renal carcinoma. Semin. Oncol. 1983, 10, 390–400. [Google Scholar]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Li, M.; Kim, W.Y. Two sides to every story: The HIF-dependent and HIF-independent functions of pVHL. J. Cell. Mol. Med. 2011, 15, 187–195. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazumder, S.; Higgins, P.J.; Samarakoon, R. Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance. Cancers 2023, 15, 1316. https://doi.org/10.3390/cancers15041316
Mazumder S, Higgins PJ, Samarakoon R. Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance. Cancers. 2023; 15(4):1316. https://doi.org/10.3390/cancers15041316
Chicago/Turabian StyleMazumder, Sonia, Paul J. Higgins, and Rohan Samarakoon. 2023. "Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance" Cancers 15, no. 4: 1316. https://doi.org/10.3390/cancers15041316
APA StyleMazumder, S., Higgins, P. J., & Samarakoon, R. (2023). Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance. Cancers, 15(4), 1316. https://doi.org/10.3390/cancers15041316