
Prognostic Value and Immune Infiltration of HPV-Related Genes in the Immune Microenvironment of Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Data Processing
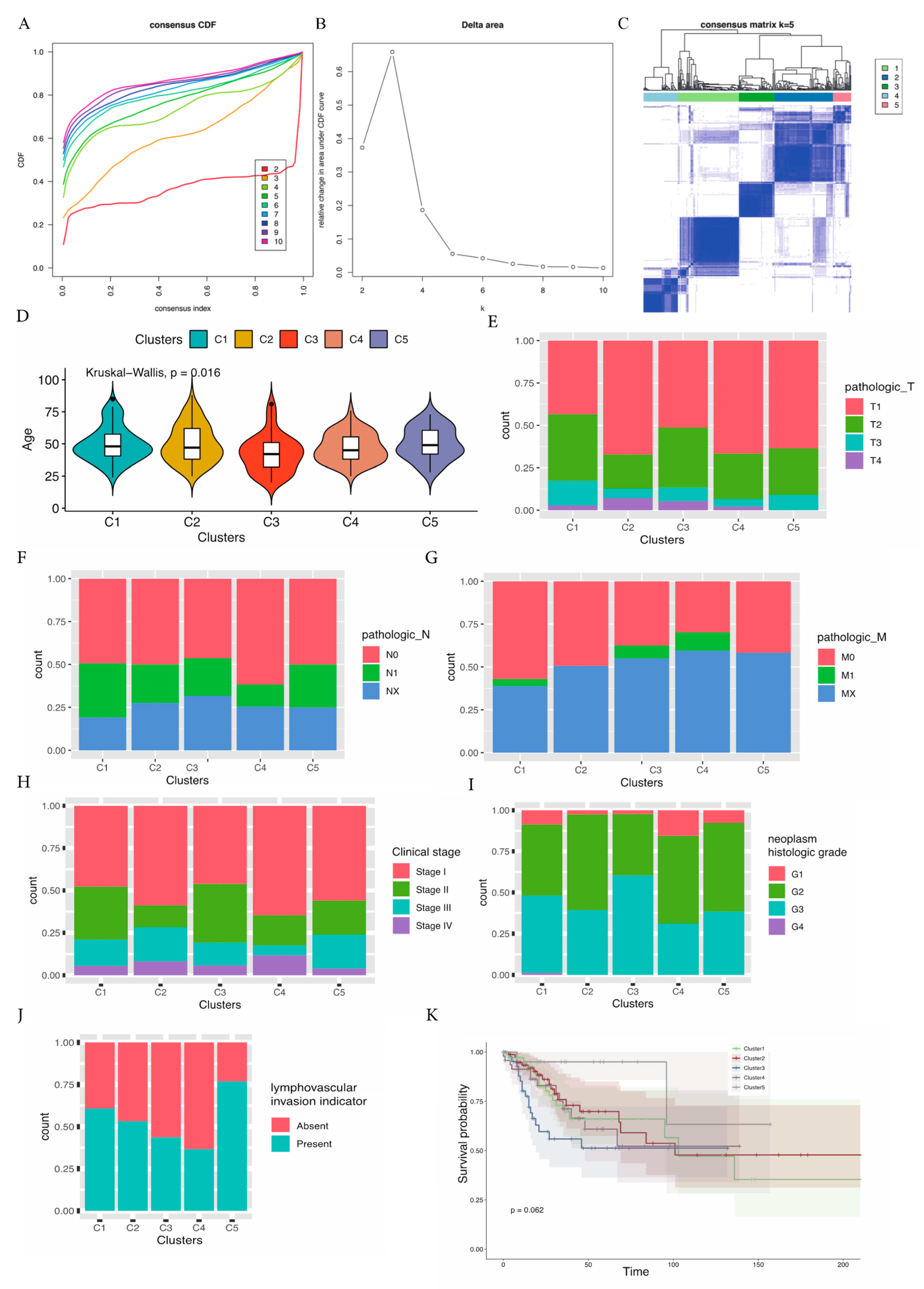
2.3. Immunogene-Based CESC Molecular Subtypes
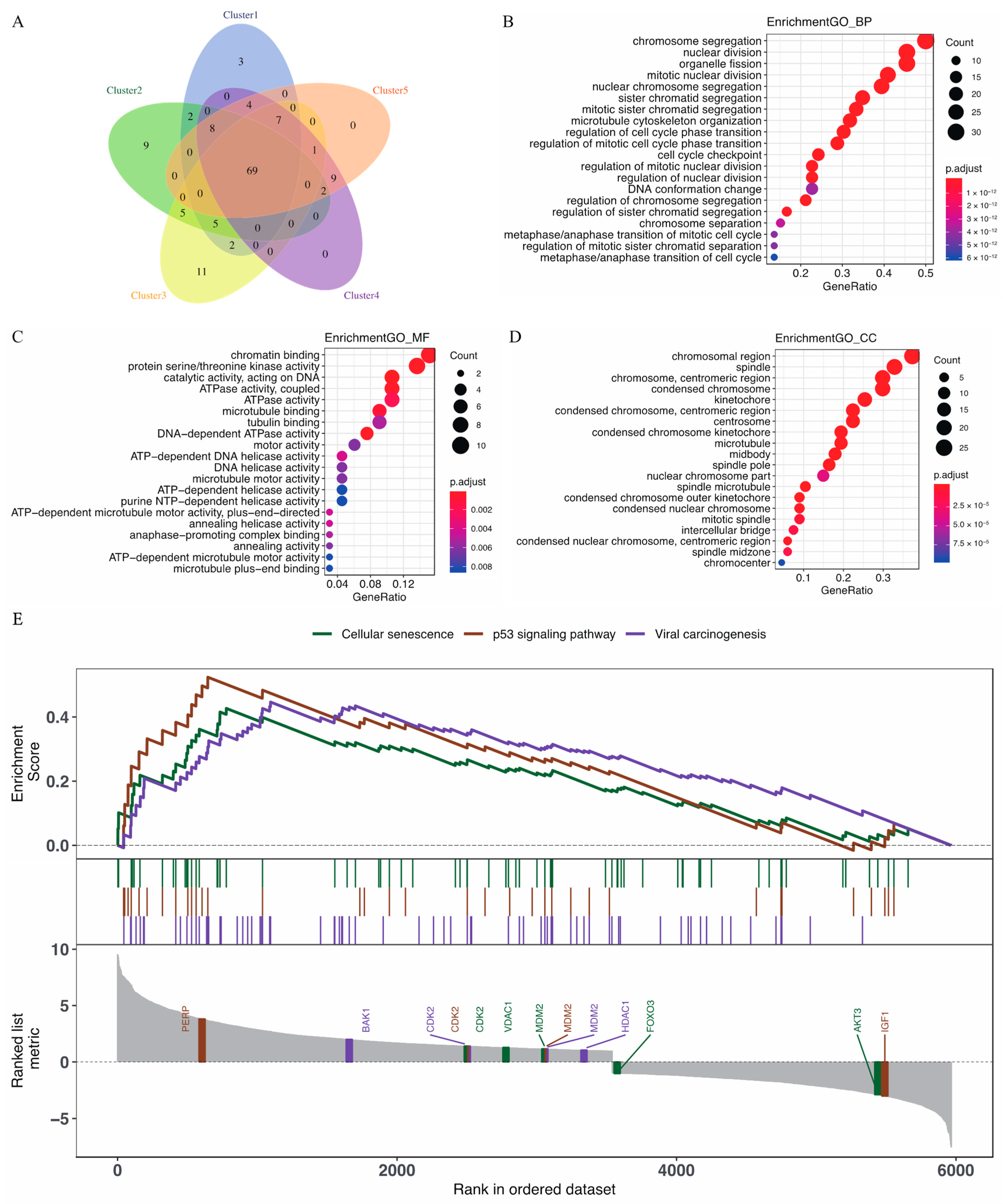
2.4. Differentially Expressed Genes
2.5. Functional Enrichment
2.6. Gene Set Enrichment Analysis
2.7. Survival Analysis
2.8. Immunohistochemistry
2.9. Statistical Analysis
3. Results
3.1. Molecular Subtypes Based on Immune Genes and Clinical Characteristics of the Different Subtypes
3.2. Estimate Scores Demonstrated a Significant Association with Clinical Outcome in Immune-Related Subtypes
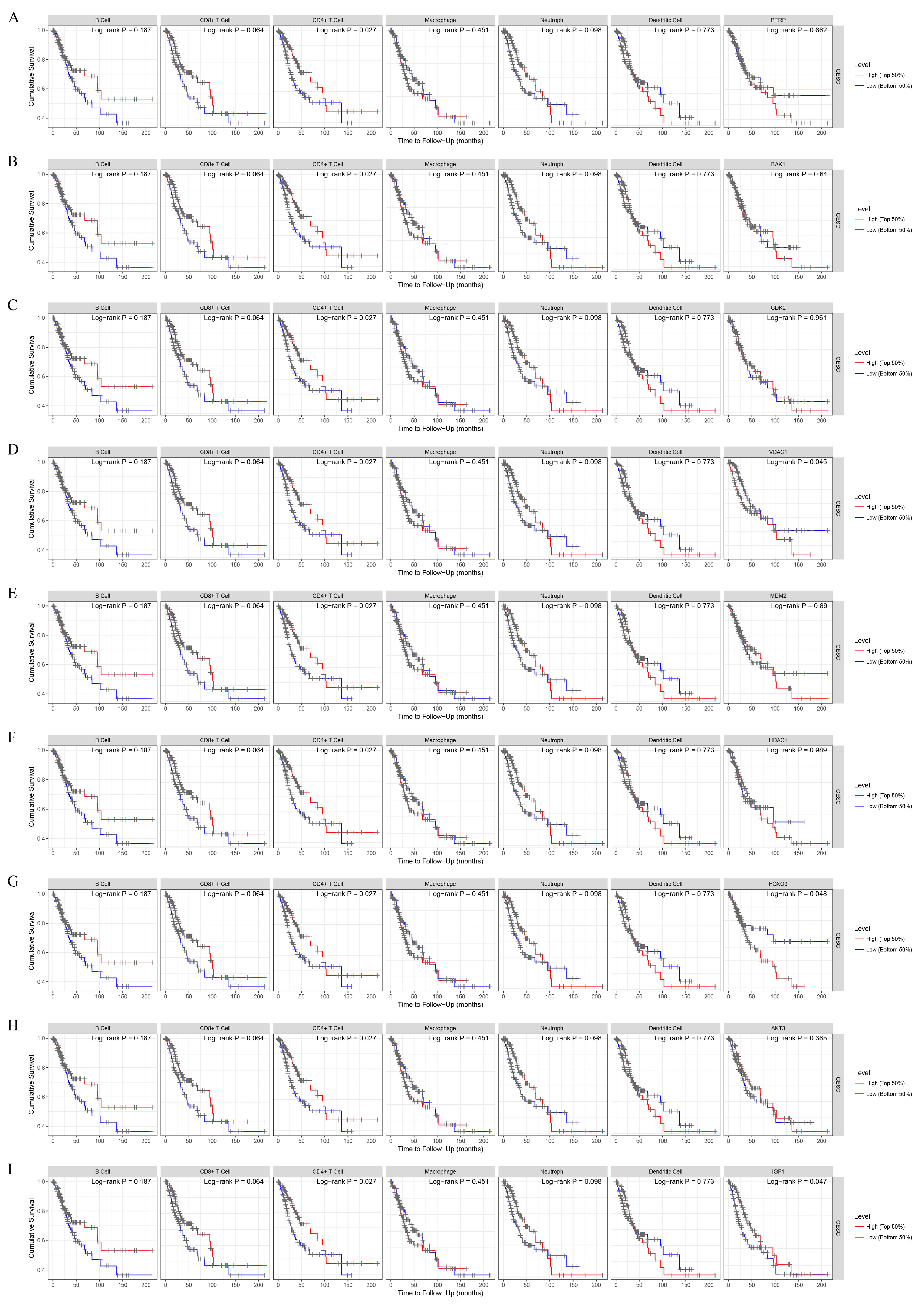
3.3. Survival Analysis of the CESC Patients in Terms of the Tumor Microenvironment for the TCGA Cohort
3.4. GO Enrichment Analysis and the Biological Pathways Identified by GSEA for Immune-Related Genes
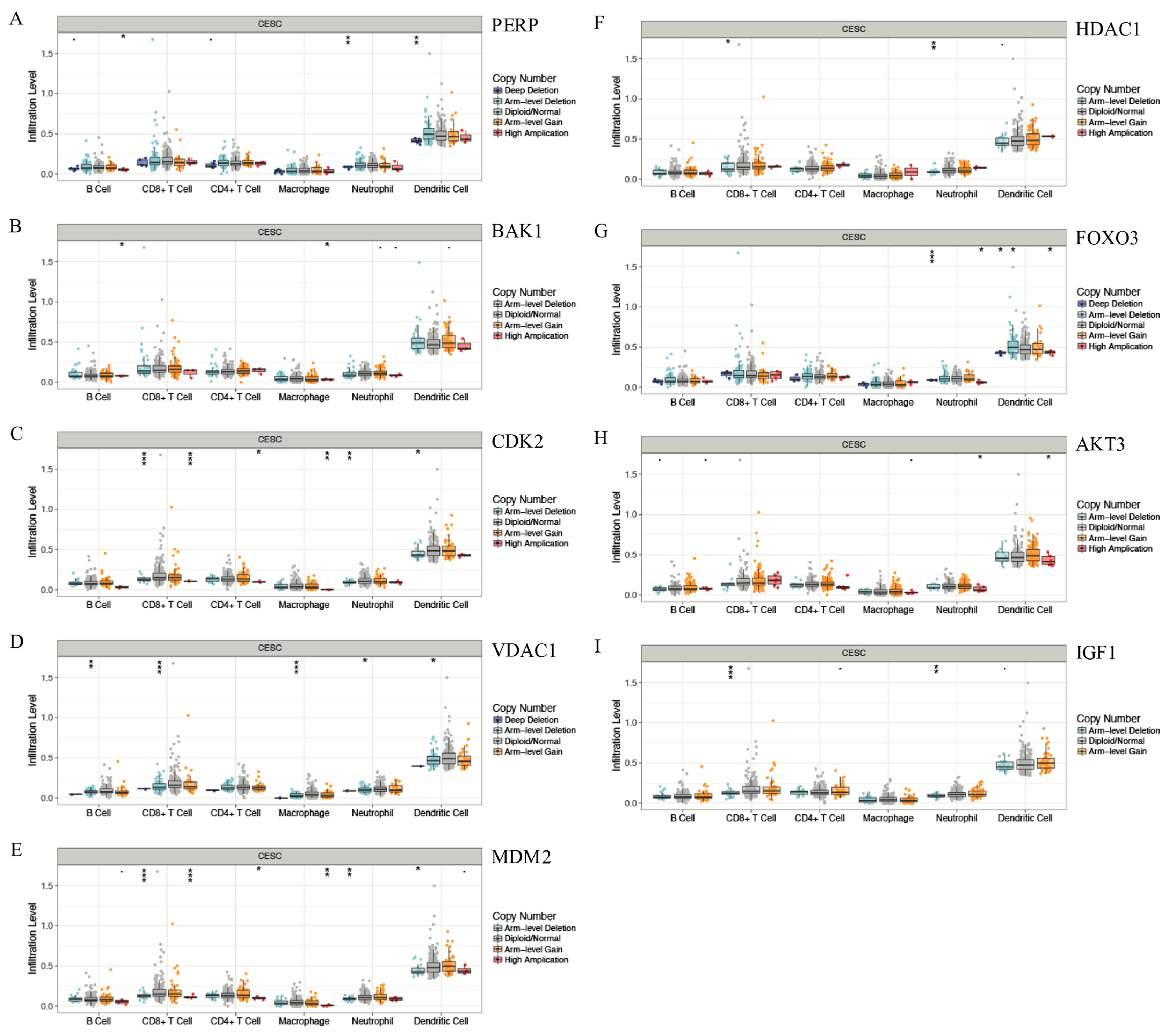
3.5. Correlation Analysis Performed on the Expression of the Key Genes and Immune Infiltration
3.6. Survival Analysis of the Expressions of the Key Genes in the Tumor Microenvironment from the TCGA Cohort
3.7. Correlation between Gene Copy Number Variation and Immune Infiltration Abundance in CESC
3.8. Correlations between the Key Genes and Prognosis in the East Hospital Cohort
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018, GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Marth, C.; Landoni, F.; Mahner, S.; McCormack, M.; Gonzalez-Martin, A.; Colombo, N. Cervical cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl 4), iv262. [Google Scholar] [CrossRef]
- Selman, T.J.; Mann, C.; Zamora, J.; Appleyard, T.L.; Khan, K. Diagnostic accuracy of tests for lymph node status in primary cervical cancer: A systematic review and meta-analysis. CMAJ 2008, 178, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wu, Y.; Deng, Y.; Zhou, L.; Yang, P.; Zheng, Y.; Zhang, D.; Zhai, Z.; Li, N.; Hao, Q.; et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1659094. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Borcoman, E.; Le Tourneau, C. Pembrolizumab in cervical cancer: Latest evidence and clinical usefulness. Ther Adv. Med. Oncol. 2017, 9, 431–439. [Google Scholar] [CrossRef]
- Chung, H.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, S.; Son, M.-J.; Kim, G.; Singh, P.; Kim, H.N.; Choi, H.-G.; Yoo, H.J.; Ko, Y.B.; Lee, B.S.; et al. Dual oxidase 1 and NADPH oxidase 2 exert favorable effects in cervical cancer patients by activating immune response. BMC Cancer 2019, 19, 1078. [Google Scholar] [CrossRef]
- Punt, S.; Houwing-Duistermaat, J.J.; Schulkens, I.A.; Thijssen, V.L.; Osse, E.M.; de Kroon, C.D.; Griffioen, A.W.; Fleuren, G.J.; Gorter, A.; Jordanova, E.S. Correlations between immune response and vascularization qRT-PCR gene expression clusters in squamous cervical cancer. Mol. Cancer 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhang, C.; Xiao, S.; Liang, L.; Liao, S.; Xiang, Y.; Cao, K.; Chen, H.; Zhou, Y. Identification of differentially expressed genes in cervical cancer by bioinformatics analysis. Oncol. Lett. 2018, 16, 2549–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Cai, Y. Methylation-Based Classification of Cervical Squamous Cell Carcinoma into Two New Subclasses Differing in Immune-Related Gene Expression. Int. J. Mol. Sci. 2018, 19, 3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Consortium, S.M.-I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.; Orris, B.; Majumdar, A.; Bhat, S.; Stivers, J.T. Macromolecular crowding induces compaction and DNA binding in the disordered N-terminal domain of hUNG2. DNA Repair. 2019, 86, 102764. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.-H.; Zhou, H.; Cooper, L.; Huang, J.-L.; Zhu, S.-B.; Zhao, X.-X.; Ding, H.; Pan, Y.-L.; Rong, L. LAYN Is a Prognostic Biomarker and Correlated With Immune Infiltrates in Gastric and Colon Cancers. Front. Immunol. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Jia, X.; Wang, C.; Xu, J.; Gao, S.J.; Lu, C. A DHX9-lncRNA-MDM2 interaction regulates cell invasion and angiogenesis of cervical cancer. Cell Death Differ. 2019, 26, 1750–1765. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [PubMed] [Green Version]
- Kim, T.; Chen, I.R.; Lin, Y.; Wang, A.Y.; Yang, J.Y.H.; Yang, P. Impact of similarity metrics on single-cell RNA-seq data clustering. Brief. Bioinform. 2018, 20, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hou, Z.; Wang, C.; Wang, H.; Zhang, H. Gene expression profiles reveal key genes for early diagnosis and treatment of adamantinomatous craniopharyngioma. Cancer Gene Ther. 2018, 25, 227–239. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Y.; Yuan, M.; Marron, J.S. Statistical Significance of Clustering using Soft Thresholding. J. Comput. Graph. Stat. 2015, 24, 975–993. [Google Scholar] [CrossRef]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, Z.; Ge, W.; Shou, J.; You, L.; Pan, H.; Han, W. Analysis of potential genes and pathways associated with the colorectal normal mucosa-adenoma-carcinoma sequence. Cancer Med. 2018, 7, 2555–2566. [Google Scholar] [CrossRef]
- Shen, Y.; Pan, X.; Yang, J. Gene regulation and prognostic indicators of lung squamous cell carcinoma: TCGA-derived miRNA/mRNA sequencing and DNA methylation data. J. Cell Physiol. 2019, 234, 22896–22910. [Google Scholar] [CrossRef]
- Willforss, J.; Chawade, A.; Levander, F. NormalyzerDE: Online Tool for Improved Normalization of Omics Expression Data and High-Sensitivity Differential Expression Analysis. J. Proteome. Res. 2019, 18, 732–740. [Google Scholar] [CrossRef]
- Ebrahimie, E.; Fruzangohar, M.; Moussavi Nik, S.H.; Newman, M. Gene Ontology-Based Analysis of Zebrafish Omics Data Using the Web Tool Comparative Gene Ontology. Zebrafish 2017, 14, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Goto, S.; Kawano, S.; Aoki-Kinoshita, K.F.; Ueda, N.; Hamajima, M.; Kawasaki, T.; Kanehisa, M. KEGG as a glycome informatics resource. Glycobiology 2006, 16, 63R–70R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, P.C.; Lee, D.S.; Fine, J.P. Introduction to the Analysis of Survival Data in the Presence of Competing Risks. Circulation 2016, 133, 601–609. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Wei, P.; Xu, Y.; Zhuo, C.; Wang, Y.; Li, D.; Cai, S. Overexpression of forkhead Box C2 promotes tumor metastasis and indicates poor prognosis in colon cancer via regulating epithelial-mesenchymal transition. Am. J. Cancer Res. 2015, 5, 2022–2034. [Google Scholar]
- Taieb, J.; Kourie, H.R.; Emile, J.-F.; Le Malicot, K.; Balogoun, R.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.-L.; Mulot, C.; et al. Association of Prognostic Value of Primary Tumor Location in Stage III Colon Cancer With RAS and BRAF Mutational Status. JAMA Oncol. 2018, 4, e173695. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef] [Green Version]
- Kenny, P.A.; Nelson, C.M.; Bissell, M.J. The Ecology of Tumors: By perturbing the microenvironment, wounds and infection may be key to tumor development. Scientist 2006, 20, 30. [Google Scholar] [PubMed]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Preprints 2022, 2022100226. [Google Scholar] [CrossRef]
- Meijer, C.; Steenbergen, R.D.M. Gynaecological cancer: Novel molecular subtypes of cervical cancer—Potential clinical consequences. Nat. Rev. Clin. Oncol. 2017, 14, 397–398. [Google Scholar] [CrossRef]
- Bai, X.; Wang, W.; Zhao, P.; Wen, J.; Guo, X.; Shen, T.; Shen, J.; Yang, X. LncRNA CRNDE acts as an oncogene in cervical cancer through sponging miR-183 to regulate CCNB1 expression. Carcinogenesis 2019, 41, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Wang, T.; Li, J.; Yan, M.; Li, D.; Li, B.; Jin, T. The impact of genetic variants in IL1R2 on cervical cancer risk among Uygur females from China: A case-control study. Mol. Genet. Genomic Med. 2019, 7, e00516. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Wu, Y.; Peng, Y.; Liu, X.; Bie, J.; Li, S. Systematic analysis to identify a key role of CDK1 in mediating gene interaction networks in cervical cancer development. Ir. J. Med. Sci. 2016, 185, 231–239. [Google Scholar] [CrossRef]
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- A Demoulin, S.; Somja, J.; Duray, A.; Guénin, S.; Roncarati, P.; O Delvenne, P.; Herfs, M.F.; Hubert, P.M. Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. Oncoimmunology 2015, 4, e1008334. [Google Scholar] [CrossRef] [Green Version]
- Mine, K.L.; Shulzhenko, N.; Yambartsev, A.; Rochman, M.; Sanson, G.F.O.; Lando, M.; Varma, S.; Skinner, J.; Volfovsky, N.; Deng, T.; et al. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat. Commun. 2013, 4, 1806. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Burg, S.H.; Piersma, S.J.; De Jong, A.; Van Der Hulst, J.M.; Kwappenberg, K.M.; Van Den Hende, M.; Welters, M.J.; Van Rood, J.J.; Fleuren, G.J.; Melief, C.J.; et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc. Natl. Acad. Sci. USA 2007, 104, 12087–12092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasper, D.J.; Tejera, M.M.; Suresh, M. CD4 T-cell memory generation and maintenance. Crit. Rev. Immunol. 2014, 34, 121–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, M.K.; Clambey, E.T.; Kappler, J.W.; Marrack, P. CD4 memory T cells: What are they and what can they do? Semin. Immunol. 2009, 21, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.C.; Mann, C.H.; Rookes, S.; Rollason, T.; Murphy, D.; Freeth, M.G.; Gallimore, P.H.; Roberts, S. T-cell responses to human papillomavirus type 16 among women with different grades of cervical neoplasia. Br. J. Cancer 2005, 93, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Monnier-Benoit, S.; Mauny, F.; Riethmuller, D.; Guerrini, J.-S.; Căpîlna, M.; Félix, S.; Seillès, E.; Mougin, C.; Prétet, J.-L. Immunohistochemical analysis of CD4+ and CD8+ T-cell subsets in high risk human papillomavirus-associated pre-malignant and malignant lesions of the uterine cervix. Gynecol. Oncol. 2006, 102, 22–31. [Google Scholar] [CrossRef]
- Lheureux, S.; Butler, M.O.; Clarke, B.; Cristea, M.C.; Martin, L.P.; Tonkin, K.; Fleming, G.F.; Tinker, A.V.; Hirte, H.W.; Tsoref, D.; et al. Association of Ipilimumab With Safety and Antitumor Activity in Women With Metastatic or Recurrent Human Papillomavirus-Related Cervical Carcinoma. JAMA Oncol. 2018, 4, e173776. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.T.; Milovanova, T.N. Mucosal Immunity and the FOXO1 Transcription Factors. Front. Immunol. 2019, 10, 2530. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.K.; Ahmad, S.M.; Idorn, M.; Met, Ö.; Martinenaite, E.; Svane, I.M.; Straten, P.T.; Andersen, M.H. Spontaneous presence of FOXO3-specific T cells in cancer patients. Oncoimmunology 2014, 3, e953411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.E.; Puccini, A.; Xiu, J.; Raghavan, D.; Lenz, H.-J.; Korn, W.M.; Shields, A.F.; Philip, P.A.; Marshall, J.L.; Goldberg, R.M. Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncologist 2018, 23, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFbeta Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, W.; Yang, X.; Wang, Z.; Hao, M. miR-375 Affects the Proliferation, Invasion, and Apoptosis of HPV16-Positive Human Cervical Cancer Cells by Targeting IGF-1R. Int. J. Gynecol. Cancer 2016, 26, 851–858. [Google Scholar] [CrossRef]
- Merritt, M.A.; Strickler, H.D.; Einstein, M.H.; Yang, H.P.; Sherman, M.E.; Wentzensen, N.; Brouwer-Visser, J.; Cossio, M.J.; Whitney, K.D.; Yu, H.; et al. Insulin/IGF and sex hormone axes in human endometrium and associations with endometrial cancer risk factors. Cancer Causes Control 2016, 27, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Pickard, A.; Durzynska, J.; McCance, D.J.; Barton, E.R. The IGF axis in HPV associated cancers. Mutat. Res. Rev. Mutat. Res. 2017, 772, 67–77. [Google Scholar] [CrossRef]
- Yeh, C.R.; Ou, Z.Y.; Xiao, G.Q.; Guancial, E.; Yeh, S. Infiltrating T cells promote renal cell carcinoma (RCC) progression via altering the estrogen receptor beta-DAB2IP signals. Oncotarget 2015, 6, 44346–44359. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Yahya, M.A.; Sharon, S.M.; Hantisteanu, S.; Hallak, M.; Bruchim, I. The Role of the Insulin-Like Growth Factor 1 Pathway in Immune Tumor Microenvironment and Its Clinical Ramifications in Gynecologic Malignancies. Front. Endocrinol. 2018, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Durzynska, J.; Barton, E. IGF expression in HPV-related and HPV-unrelated human cancer cells. Oncol. Rep. 2014, 32, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, J.; Shi, X.; Li, S.; Tang, P.M.K.; Li, Z.; Li, H.; Wei, C. Antimalarial Dihydroartemisinin triggers autophagy within HeLa cells of human cervical cancer through Bcl-2 phosphorylation at Ser70. Phytomedicine 2019, 52, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ming, P.; Cai, T.; Li, J.; Ning, Y.; Xie, S.; Tao, T.; Tang, F. A novel arylbenzofuran induces cervical cancer cell apoptosis and G1/S arrest through ERK-mediated Cdk2/cyclin-A signaling pathway. Oncotarget 2016, 7, 41843–41856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ding, W.; Liu, Y.; Hu, Z.; Zhu, D.; Wang, X.; Yu, L.; Wang, L.; Shen, H.; Zhang, W.; et al. Proteomics-based identification of VDAC1 as a tumor promoter in cervical carcinoma. Oncotarget 2016, 7, 52317–52328. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, L.; Chen, Z. Transcriptome profiling of cervical cancer cells acquired resistance to cisplatin by deep sequencing. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2820–2829. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Luo, Z.; Li, Z.; Liu, Y.; Zhao, Q. PERP gene therapy attenuates lung cancer xenograft via inducing apoptosis and suppressing VEGF. Cancer Biol. Ther. 2011, 12, 1114–1119. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | No. (%) | FOXO3 Expression | χ2 | Log-Rank-p No. (%) | No. (%) | IGF1 Expression | χ2 | Log-Rank-p No. (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Low (%) | High (%) | Low (%) | High (%) | |||||||
| Age | 11.989 | <0.001 | 13.123 | <0.001 | ||||||
| <50 | 43 (75.44) | 22 (75.86) | 21 (75.00) | 43 (75.44) | 20 (76.92) | 23 (74.19) | ||||
| ≥50 | 14 (24.56) | 7 (24.14) | 7 (25.00) | 14 (24.56) | 6 (23.08) | 8 (25.81) | ||||
| T category | 14.746 | <0.001 | 12.926 | 0.002 | ||||||
| 1 | 31 (54.39) | 17 (58.62) | 14 (50.00) | 31 (54.39) | 12 (46.15) | 19 (61.29) | ||||
| 2 | 14 (24.56) | 6 (20.69) | 8 (28.57) | 14 (24.56) | 5 (19.23) | 9 (29.03) | ||||
| 3/4 | 12 (21.05) | 6 (20.69) | 6 (21.43) | 12 (21.05) | 9 (34.62) | 3 (9.68) | ||||
| N stage | 15.670 | <0.001 | 11.657 | <0.001 | ||||||
| 0 | 46 (80.70) | 23 (79.31) | 23 (82.14) | 46 (80.70) | 17 (65.38) | 29 (93.55) | ||||
| 1 | 11 (19.30) | 6 (20.69) | 5 (17.86) | 11 (19.30) | 9 (34.62) | 2 (6.45) | ||||
| Pathology grade | 0.930 | 0.335 | 0.894 | 0.344 | ||||||
| I–II | 16 (23.53) | 8 (27.59) | 6 (21.43) | 14 (24.56) | 5 (19.23) | 9 (26.03) | ||||
| III | 52 (76.47) | 21 (72.41) | 22 (78.57) | 43 (75.44) | 21 (80.77) | 22 (70.97) | ||||
| HPV infection | 0.393 | 0.531 | 0.673 | 0.412 | ||||||
| Negative | 10 (17.54) | 6 (20.69) | 4 (14.29) | 10 (17.54) | 6 (23.08) | 4 (12.90) | ||||
| Positive | 47 (82.46) | 23 (79.31) | 24 (85.71) | 47 (82.46) | 20 (76.92) | 27 (87.10) | ||||
| Recidivation | 52.145 | <0.001 | 60.813 | <0.001 | ||||||
| Negative | 44 (77.19) | 25 (86.21) | 19 (67.86) | 44 (77.19) | 20 (76.92) | 24 (77.42) | ||||
| Positive | 13 (22.81) | 4 (13.79) | 9 (32.14) | 13 (22.81) | 6 (23.08) | 7 (22.58) | ||||
| IGF1 expression | 1.138 | 0.286 | 1.700 | 0.192 | ||||||
| Low | 26 (45.61) | 17 (58.62) | 9 (32.14) | 29 (50.88) | 17 (65.38) | 12 (38.71) | ||||
| High | 31 (54.39) | 12 (41.38) | 19 (67.86) | 28 (49.12) | 9 (34.62) | 19 (61.29) | ||||
| Factor | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | |
| Age | 6.101 (2.724, 13.67) | <0.001 | 7.959 (1.742, 36.364) | 0.007 |
| T category | 12.73 (6.073, 26.67) | <0.001 | 16.414 (0.844, 319.14) | 0.065 |
| N stage | 4.658 (1.659, 13.08) | <0.001 | 6.892 (1.512, 31.408) | 0.013 |
| Pathology grade | 1.149 (0.5268, 2.504) | 0.7346 | 0.296 (0.057, 1.523) | 0.145 |
| HPV infection | 1.016 (0.3537, 2.921) | 0.9759 | 5.447 (0.430, 68.967) | 0.191 |
| IGF1 | 0.454 (0.208, 0.989) | 0.0438 | 0.090 (0.008, 1.081) | 0.058 |
| FOXO3 | 2.473 (1.046, 5.845) | 0.0423 | 11.611 (1.033, 130.506) | 0.047 |
| BAK | 0.872 (0.439, 1.731) | 0.6924 | 0.817 (0.081, 8.286) | 0.864 |
| VDAC1 | 1.377 (0.680, 2.787) | 0.3943 | 0.439 (0.069, 2.784) | 0.382 |
| AKT3 | 1.435 (0.701, 2.936) | 0.3282 | 12.481 (0.214, 729.339) | 0.357 |
| PERP | 0.855 (0.393, 1.862) | 0.6892 | 0.515 (0.143, 2.650) | 0.515 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, Q.; Mao, L.; Shi, R.; Chang, L.; Wang, G.; Cheng, J.; Chen, R. Prognostic Value and Immune Infiltration of HPV-Related Genes in the Immune Microenvironment of Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma. Cancers 2023, 15, 1419. https://doi.org/10.3390/cancers15051419
Gan Q, Mao L, Shi R, Chang L, Wang G, Cheng J, Chen R. Prognostic Value and Immune Infiltration of HPV-Related Genes in the Immune Microenvironment of Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma. Cancers. 2023; 15(5):1419. https://doi.org/10.3390/cancers15051419
Chicago/Turabian StyleGan, Qiyu, Luning Mao, Rui Shi, Linlin Chang, Guozeng Wang, Jingxin Cheng, and Rui Chen. 2023. "Prognostic Value and Immune Infiltration of HPV-Related Genes in the Immune Microenvironment of Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma" Cancers 15, no. 5: 1419. https://doi.org/10.3390/cancers15051419
APA StyleGan, Q., Mao, L., Shi, R., Chang, L., Wang, G., Cheng, J., & Chen, R. (2023). Prognostic Value and Immune Infiltration of HPV-Related Genes in the Immune Microenvironment of Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma. Cancers, 15(5), 1419. https://doi.org/10.3390/cancers15051419