

Genomics-Driven Precision Medicine in Pediatric Solid Tumors

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
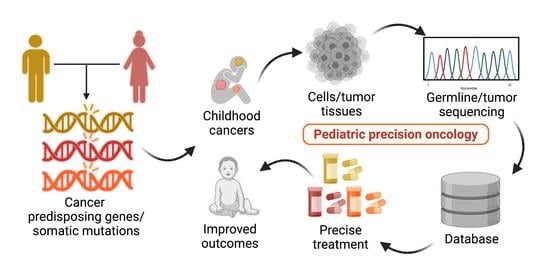
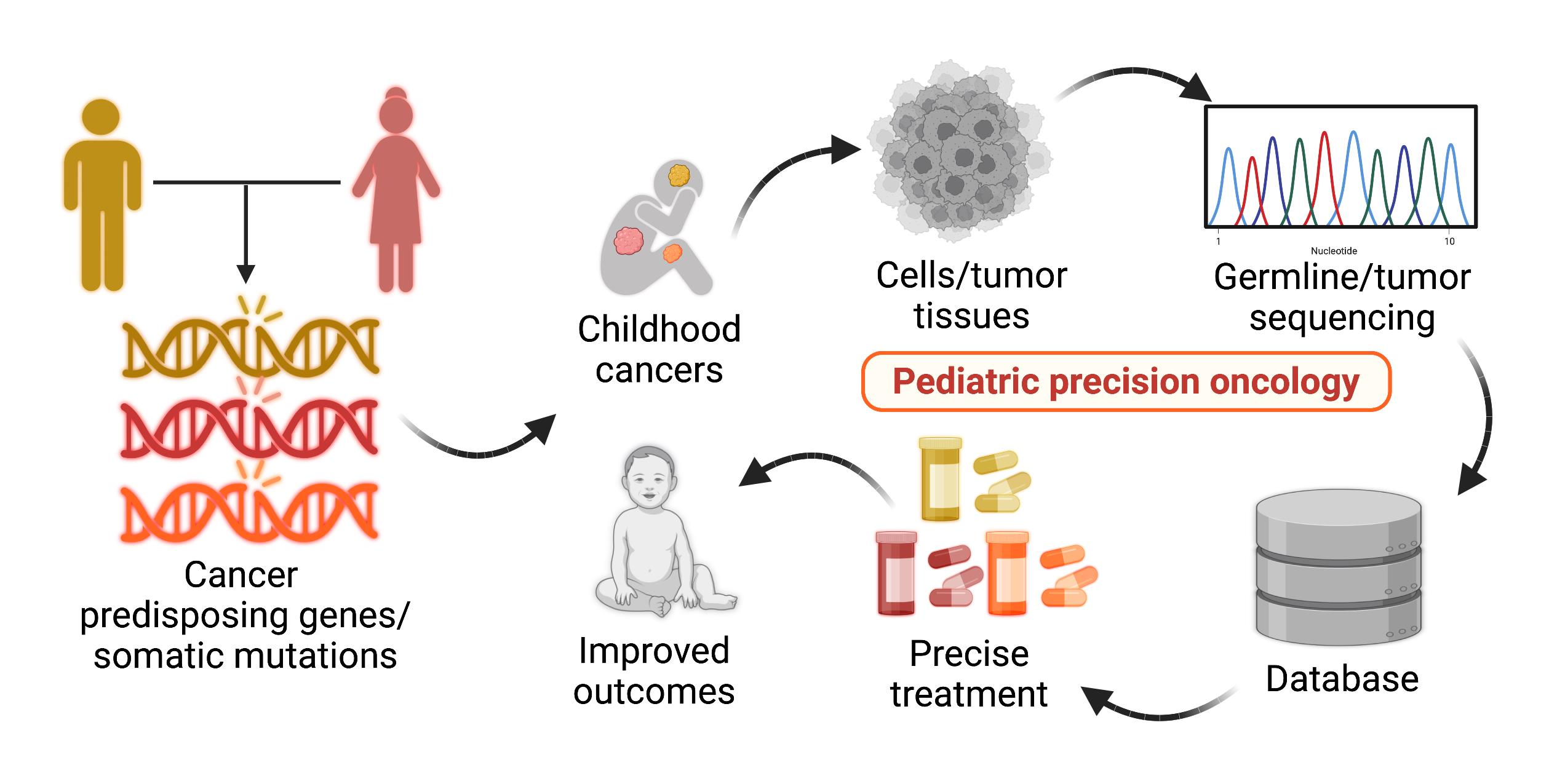
1. Introduction
2. Genetic Alterations on Cancer Hallmarks
2.1. Cancer Hallmarks and Common Targeted Signaling Pathways
2.2. Tumor Cells Have Both Germline and Somatic Variants in Their Genome
- Familial history of the same or related cancers;
- Occurrence of bilateral or multifocal cancers;
- Earlier age at disease onset;
- Physical suggestive of a predisposition syndrome;
- Appearance of specific tumor types corresponding to the genetic predisposition.
2.3. Germline and Somatic Variants Classified as Druggable
3. Pediatric Cancer Genome
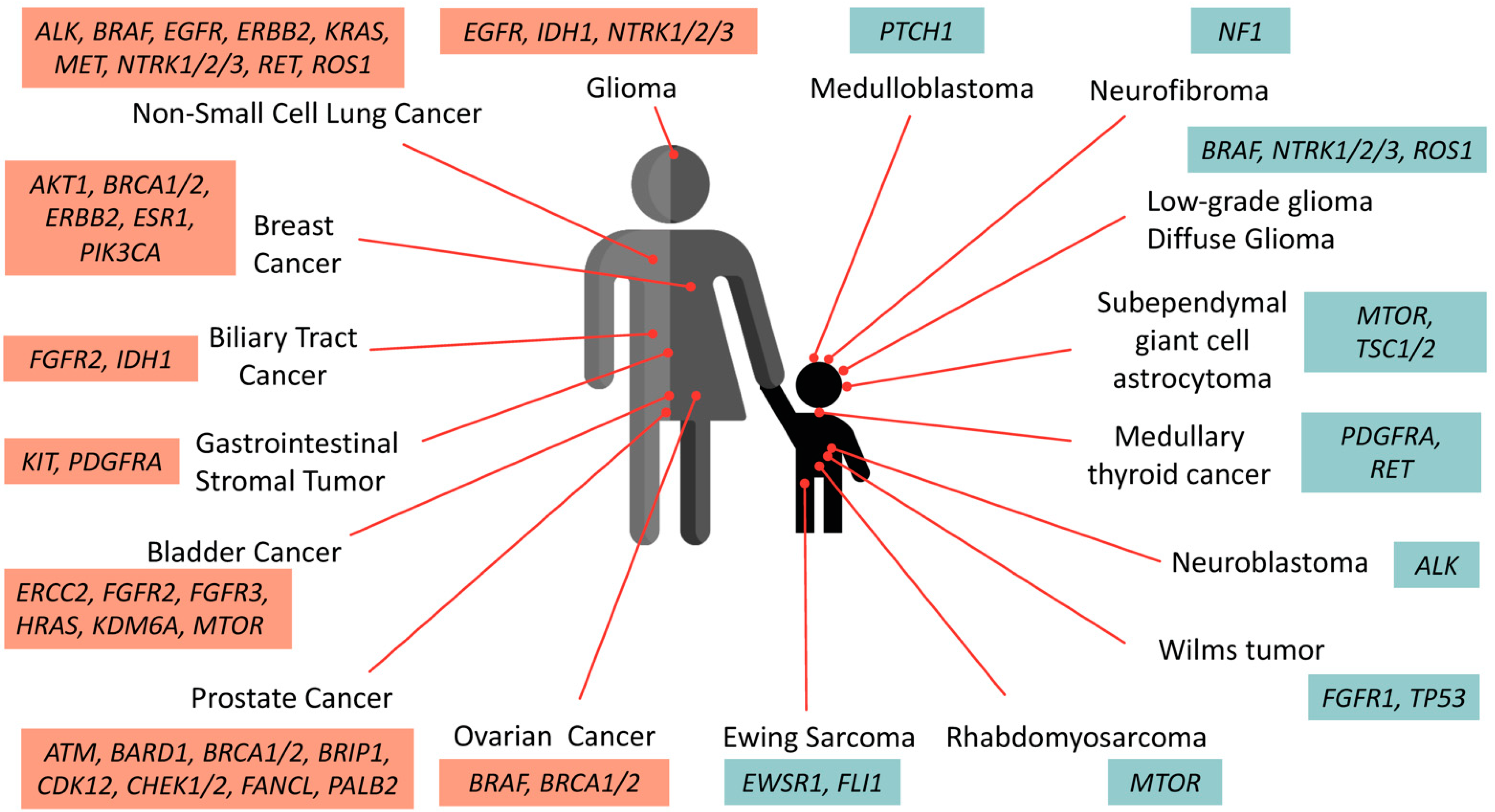
3.1. Pediatric vs. Adult Cancer Development
3.2. Somatic and Germline Mutations Identified in Pediatric Cancer Cohorts
3.3. Predictive and Common Genetic Variant Abnormalities Identified in Pediatric Tumors
4. Current Progress in Clinical Trials for Pediatric Precision Oncology
5. Challenges and Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TRK | Tyrosine receptor kinase |
| MAPK | Mitogen-activating protein kinase |
| PI3K | Phosphoinositide 3-kinases |
| NTRK | Neurotrophic tyrosine receptor kinase |
| CPG | Cancer predisposing gene |
| CNS | Central nervous system |
| RTK | Receptor tyrosine kinase |
| WGS | Whole genome sequencing |
| WES | Whole exome sequencing |
| TGFB | Transforming growth factor beta |
| NSCLC | Non-small cell lung cancer |
References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 Contributors. International incidence of childhood cancer, 2001–10: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tsui, P.C.; Lee, Y.-F.; Liu, Z.W.Y.; Ip, L.R.H.; Piao, W.; Chiang, A.K.S.; Lui, V.W.Y. An update on genomic-guided therapies for pediatric solid tumors. Future Oncol. 2017, 13, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, P.; Ortiz, R.; Fuentes, S.; Gamboa, Y.; Ah Chu-Sanchez, M.S.; Arambú, I.C.; Montero, M.; Báez, F.; Rodríguez-Galindo, C.; Antillón-Klussmann, F.; et al. Barriers to effective treatment of pediatric solid tumors in middle-income countries: Can we make sense of the spectrum of nonbiologic factors that influence outcomes? Cancer 2014, 120, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Lupo, P.J.; Spector, L.G. Cancer Progress and Priorities: Childhood Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1081–1094. [Google Scholar] [CrossRef]
- Langenberg, K.P.S.; Looze, E.J.; Molenaar, J.J. The Landscape of Pediatric Precision Oncology: Program Design, Actionable Alterations, and Clinical Trial Development. Cancers 2021, 13, 4324. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Vundamati, D.S.; Farooqi, M.S.; Guest, E. Precision Medicine in Pediatric Cancer: Current Applications and Future Prospects. High-Throughput 2018, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 160–169. [Google Scholar] [CrossRef]
- Butler, E.; Ludwig, K.; Pacenta, H.L.; Klesse, L.J.; Watt, T.C.; Laetsch, T.W. Recent progress in the treatment of cancer in children. CA A Cancer J. Clin. 2021, 71, 315–332. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Chinnaiyan, A.M. Precision oncology in the age of integrative genomics. Nat. Biotechnol. 2018, 36, 46–60. [Google Scholar] [CrossRef]
- Abrams, J.; Conley, B.; Mooney, M.; Zwiebel, J.; Chen, A.; Welch, J.J.; Takebe, N.; Malik, S.; McShane, L.; Korn, E.; et al. National Cancer Institute’s Precision Medicine Initiatives for the New National Clinical Trials Network. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, 71–76. [Google Scholar] [CrossRef]
- Wong, M.; Mayoh, C.; Lau, L.M.S.; Khuong-Quang, D.A.; Pinese, M.; Kumar, A.; Barahona, P.; Wilkie, E.E.; Sullivan, P.; Bowen-James, R.; et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 2020, 26, 1742–1753. [Google Scholar] [CrossRef]
- Worst, B.C.; Van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.A.-O.X.; Corson, L.A.-O.; Stegmaier, K.; Janeway, K.A.-O. Ushering in the next generation of precision trials for pediatric cancer. Science 2019, 15, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Vassal, G.; Geoerger, B.; Morland, B. Is the European Pediatric Medicine Regulation Working for Children and Adolescents with Cancer? Clin. Cancer Res. 2013, 19, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Nishiwaki, S.; Ando, Y. Gap between pediatric and adult approvals of molecular targeted drugs. Sci. Rep. 2020, 10, 17145. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib inTRKFusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Robinson, G.W.; Gajjar, A.J.; Gauvain, K.M.; Basu, E.M.; Macy, M.E.; Maese, L.D.; Sabnis, A.J.; Foster, J.H.; Shusterman, S.; Yoon, J.; et al. Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. J. Clin. Oncol. 2019, 37, 10009. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Chiangjong, W.; Chutipongtanate, S. EV-out or EV-in: Tackling cell-to-cell communication within the tumor microenvironment to enhance anti-tumor efficacy using extracellular vesicle-based therapeutic strategies. OpenNano 2022, 8, 100085. [Google Scholar] [CrossRef]
- Csermely, P.; Korcsmáros, T.; Nussinov, R. Intracellular and intercellular signaling networks in cancer initiation, development and precision anti-cancer therapy. Semin. Cell Dev. Biol. 2016, 58, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Dancey, J.E.; Bedard, P.L.; Onetto, N.; Hudson, T.J. The Genetic Basis for Cancer Treatment Decisions. Cell 2012, 148, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; Loeffen, J.L.C.M.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.L.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children with Solid Tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Scollon, S.; Anglin, A.K.; Thomas, M.; Turner, J.T.; Wolfe Schneider, K. A Comprehensive Review of Pediatric Tumors and Associated Cancer Predisposition Syndromes. J. Genet. Couns. 2017, 26, 387–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripperger, T.; Bielack, S.S.; Borkhardt, A.; Brecht, I.B.; Burkhardt, B.; Calaminus, G.; Debatin, K.-M.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes—A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med. Genet. Part A 2017, 173, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Coury, S.A.; Schneider, K.A.; Schienda, J.; Tan, W.H. Recognizing and Managing Children with a Pediatric Cancer Predisposition Syndrome: A Guide for the Pediatrician. Pediatr. Ann. 2018, 47, e204–e216. [Google Scholar] [CrossRef]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.N.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Nichols, K.E.; Plon, S.E.; Schiffman, J.D.; Malkin, D. Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin. Cancer Res. 2017, 23, e1–e5. [Google Scholar] [CrossRef] [Green Version]
- Lindor, N.M.; McMaster, M.L.; Lindor, C.J.; Greene, M.H. Concise Handbook of Familial Cancer Susceptibility Syndromes—Second Edition. JNCI Monogr. 2008, 2008, 3–93. [Google Scholar] [CrossRef] [Green Version]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin. Cancer Res. 2017, 23, e115–e122. [Google Scholar] [CrossRef] [Green Version]
- Tabori, U.; Hansford, J.R.; Achatz, M.I.; Kratz, C.P.; Plon, S.E.; Frebourg, T.; Brugières, L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin. Cancer Res. 2017, 23, e32–e37. [Google Scholar] [CrossRef] [Green Version]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mossé, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, C.P.; Achatz, M.I.; Brugières, L.; Frebourg, T.; Garber, J.E.; Greer, M.-L.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumamoto, T.; Yamazaki, F.; Nakano, Y.; Tamura, C.; Tashiro, S.; Hattori, H.; Nakagawara, A.; Tsunematsu, Y. Medical guidelines for Li–Fraumeni syndrome 2019, version 1.1. Int. J. Clin. Oncol. 2021, 26, 2161–2178. [Google Scholar] [CrossRef]
- Ahlawat, S.; Blakeley, J.O.; Langmead, S.; Belzberg, A.J.; Fayad, L.M. Current status and recommendations for imaging in neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Skelet. Radiol. 2020, 49, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Kamihara, J.; Evans, D.G.R.; Brugières, L.; Bourdeaut, F.; Molenaar, J.J.; Walsh, M.F.; Brodeur, G.M.; Diller, L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin. Cancer Res. 2017, 23, e62–e67. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, J.D.; Tomlinson, G.E.; Druker, H.; Kamihara, J.; Kohlmann, W.K.; Kratz, C.P.; Nathanson, K.L.; Pajtler, K.W.; Parareda, A.; Rednam, S.P.; et al. Multiple Endocrine Neoplasia and Hyperparathyroid-Jaw Tumor Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e123–e132. [Google Scholar] [CrossRef] [Green Version]
- Achatz, M.I.; Porter, C.C.; Brugières, L.; Druker, H.; Frebourg, T.; Foulkes, W.D.; Kratz, C.P.; Kuiper, R.P.; Hansford, J.R.; Hernandez, H.S.; et al. Cancer Screening Recommendations and Clinical Management of Inherited Gastrointestinal Cancer Syndromes in Childhood. Clin. Cancer Res. 2017, 23, e107–e114. [Google Scholar] [CrossRef] [Green Version]
- Northrup, H.; Koenig, M.K.; Pearson, D.A.; Au, K.S. Tuberous Sclerosis Complex. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jongmans, M.C.J.; van der Burgt, I.; Hoogerbrugge, P.M.; Noordam, K.; Yntema, H.G.; Nillesen, W.M.; Kuiper, R.P.; Ligtenberg, M.J.L.; van Kessel, A.G.; van Krieken, J.H.J.M.; et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 2011, 19, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Minati, R.; Perreault, C.; Thibault, P. A Roadmap Toward the Definition of Actionable Tumor-Specific Antigens. Front. Immunol. 2020, 11, 583287. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by Gene Amplification or by Splice Mutations Deleting the Juxtamembrane Domain in Primary Resected Lung Cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658. [Google Scholar] [CrossRef]
- Carr, T.H.; McEwen, R.; Dougherty, B.; Johnson, J.H.; Dry, J.R.; Lai, Z.; Ghazoui, Z.; Laing, N.M.; Hodgson, D.R.; Cruzalegui, F.; et al. Defining actionable mutations for oncology therapeutic development. Nat. Rev. Cancer 2016, 16, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.H.; Axtmayer, J.; Gustin, J.P.; Rajpurohit, A.; Lauring, J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: Implications for personalized medicine. Oncotarget 2013, 4, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine. Childhood Cancer and Functional Impacts across the Care Continuum; The National Academies Press: Washington, DC, USA, 2021; p. 526. [Google Scholar]
- Ferrari, A.; Casanova, M.; Massimino, M.; Sultan, I. Peculiar features and tailored management of adult cancers occurring in pediatric age. Expert Rev. Anticancer Ther. 2010, 10, 1837–1851. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Banito, A.; Grünewald, T.G.P.; Haber, M.; Jäger, N.; Kool, M.; Milde, T.; Molenaar, J.J.; Nabbi, A.; Pugh, T.J.; et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat. Rev. Cancer 2019, 19, 420–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahal, Z.; Abdulhai, F.; Kadara, H.; Saab, R. Genomics of adult and pediatric solid tumors. Am. J. Cancer Res. 2018, 8, 1356–1386. [Google Scholar] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.H.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.G.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Diets, I.J.; Waanders, E.; Ligtenberg, M.J.; van Bladel, D.A.G.; Kamping, E.J.; Hoogerbrugge, P.M.; Hopman, S.; Olderode-Berends, M.J.; Gerkes, E.H.; Koolen, D.A.; et al. High Yield of Pathogenic Germline Mutations Causative or Likely Causative of the Cancer Phenotype in Selected Children with Cancer. Clin. Cancer Res. 2018, 24, 1594–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, C.; Gout, A.M.; Zhou, X.; Thrasher, A.; Rahbarinia, D.; Brady, S.W.; Macias, M.; Birch, K.; Finkelstein, D.; Sunny, J.; et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 2021, 11, 1082–1099. [Google Scholar] [CrossRef]
- Krysiak, K.; Danos, A.M.; Saliba, J.; McMichael, J.F.; Coffman, A.C.; Kiwala, S.; Barnell, E.K.; Sheta, L.; Grisdale, C.J.; Kujan, L.; et al. CIViCdb 2022: Evolution of an open-access cancer variant interpretation knowledgebase. Nucleic Acids Res. 2022, 51, D1230–D1241. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönherr, C.; Ruuth, K.; Yamazaki, Y.; Eriksson, T.; Christensen, J.; Palmer, R.H.; Hallberg, B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem. J. 2011, 440, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, S.G.; Laetsch, T.W.; Federman, N.; Turpin, B.K.; Albert, C.M.; Nagasubramanian, R.; Anderson, M.E.; Davis, J.L.; Qamoos, H.E.; Reynolds, M.E.; et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 2018, 124, 4241–4247. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Dubois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Mangum, R.; Reuther, J.; Bertrand, K.C.; Chandramohan, R.; Kukreja, M.K.; Paulino, A.C.; Muzny, D.; Hu, J.; Gibbs, R.A.; Curry, D.J.; et al. Durable Response to Larotrectinib in a Child with Histologic Diagnosis of Recurrent Disseminated Ependymoma Discovered to Harbor an NTRK2 Fusion: The Impact of Integrated Genomic Profiling. JCO Precis. Oncol. 2021, 5, 1221–1227. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.; Baldwin, A.; Dombi, E.; Fisher, M.J.; Weiss, B.D.; Kim, A.; Blakeley, J.O.N.; Whitcomb, P.; Holmblad, M.; et al. SPRINT: Phase II study of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886) in children with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). J. Clin. Oncol. 2018, 36, 10503. [Google Scholar] [CrossRef]
- Subbiah, V.; Westin, S.N.; Wang, K.; Araujo, D.; Wang, W.-L.; Miller, V.A.; Ross, J.S.; Stephens, P.J.; Palmer, G.A.; Ali, S.M. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J. Hematol. Oncol. 2014, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Del Bufalo, F.; Carai, A.; Figà-Talamanca, L.; Pettorini, B.; Mallucci, C.; Giangaspero, F.; Antonelli, M.; Badiali, M.; Moi, L.; Bianco, G.; et al. Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J. Transl. Med. 2014, 12, 356. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, D.R.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.J.; Hansford, J.R.; Geoerger, B.; Hingorani, P.; Dunkel, I.J.; Russo, M.W.; et al. Efficacy and Safety of Dabrafenib in Pediatric Patients with BRAF V600 Mutation–Positive Relapsed or Refractory Low-Grade Glioma: Results from a Phase I/IIa Study. Clin. Cancer Res. 2019, 25, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Khamaysi, Z.; Bochner, R.; Indelman, M.; Magal, L.; Avitan-Hersh, E.; Sarig, O.; Sprecher, E.; Bergman, R. Segmental basal cell naevus syndrome caused by an activating mutation in smoothened. Br. J. Dermatol. 2016, 175, 178–181. [Google Scholar] [CrossRef]
- Capone, S.; Ketonen, L.; Weathers, S.P.; Subbiah, V. Activity of Pemigatinib in Pilocytic Astrocytoma and FGFR1(N546K) Mutation. JCO Precis. Oncol. 2022, 6, e2100371. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results from Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Lv, S.; Teugels, E.; Sadones, J.A.N.; Quartier, E.; Huylebrouck, M.; Du Four, S.; Le Mercier, M.; De Witte, O.; Salmon, I.; Michotte, A.; et al. Correlation between IDH1 Gene Mutation Status and Survival of Patients Treated for Recurrent Glioma. Anticancer Res. 2011, 31, 4457. [Google Scholar]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef]
- Bresler Scott, C.; Wood Andrew, C.; Haglund Elizabeth, A.; Courtright, J.; Belcastro Lili, T.; Plegaria Jefferson, S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter Erica, L.; et al. Differential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants Found in Neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor–Naive ALK-Positive Non–Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. J. Clin. Oncol. 2020, 38, 3592–3603. [Google Scholar] [CrossRef]
- Shaw, A.T.; Bauer, T.M.; De Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent—Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.W.; Orr, B.A.; Gajjar, A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014, 14, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Silva, A.; Han, H.J.; Lang, S.-S.; Zhu, Y.; Boucher, K.; Smith, T.E.; Vakil, A.; Diviney, P.; Choudhari, N.; et al. Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 84697–84713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Baghdadi, T.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Rich, P.; Ahn, E.R.; Chai, S.; Rygiel, A.L.; Osayameh, O.; Antonelli, K.R.; et al. Sunitinib in Patients with Metastatic Colorectal Cancer (mCRC) with FLT-3 Amplification: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.G.; Tait, D.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Schink, J.C.; Alvarez, R.H.; Veljovich, D.; Cannon, T.L.; Crilley, P.A.; et al. Cetuximab in Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Ovarian Cancer without KRAS, NRAS, or BRAF Mutations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.S.; Mangat, P.K.; Garrett-Mayer, E.; Halabi, S.; Hansra, D.; Calfa, C.J.; Khalil, M.F.; Ahn, E.R.; Cannon, T.L.; Crilley, P.; et al. Pembrolizumab in Patients with Metastatic Breast Cancer with High Tumor Mutational Burden: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. J. Clin. Oncol. 2021, 39, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Janeway, K.A.; Patton, D.R.; Winter, C.L.; Coffey, B.; Williams, P.M.; Roy-Chowdhuri, S.; Tsongalis, G.J.; Routbort, M.; Ramirez, N.C.; et al. Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients with Refractory Cancers in the National Cancer Institute–Children’s Oncology Group Pediatric MATCH Trial. J. Clin. Oncol. 2022, 40, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.M.; Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.-H.; Ding, L.; Ley, T.J.; Evans, W.E. The Pediatric Cancer Genome Project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef] [Green Version]
- Sposto, R.; Stram, D.O. A strategic view of randomized trial design in low-incidence paediatric cancer. Stat. Med. 1999, 18, 1183–1197. [Google Scholar] [CrossRef]
- Deley, M.C.; Ballman, K.V.; Marandet, J.; Sargent, D. Taking the long view: How to design a series of Phase III trials to maximize cumulative therapeutic benefit. Clin. Trials 2012, 9, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too many targets, not enough patients: Rethinking neuroblastoma clinical trials. Nat. Rev. Cancer 2018, 18, 389–400. [Google Scholar] [CrossRef]
- Renfro, L.A.; Ji, L.; Piao, J.; Onar-Thomas, A.; Kairalla, J.A.; Alonzo, T.A. Trial Design Challenges and Approaches for Precision Oncology in Rare Tumors: Experiences of the Children’s Oncology Group. JCO Precis. Oncol. 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, W. Precision medicine becomes reality-tumor type-agnostic therapy. Cancer Commun. 2018, 38, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Califano, A.; Alvarez, M.J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 2017, 17, 116–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, S.; Meller, J.; Hongeng, S.; Tohtong, R.; Chutipongtanate, S. Transcriptional Regulation of Cancer Immune Checkpoints: Emerging Strategies for Immunotherapy. Vaccines 2020, 8, 735. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Pomella, S.; Rota, R. The CRISP(Y) Future of Pediatric Soft Tissue Sarcomas. Front. Chem. 2020, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ohmura, S.; Marchetto, A.; Orth, M.F.; Imle, R.; Dallmayer, M.; Musa, J.; Knott, M.M.L.; Holting, T.L.B.; Stein, S.; et al. Therapeutic targeting of the PLK1-PRC1-axis triggers cell death in genomically silent childhood cancer. Nat. Commun. 2021, 12, 5356. [Google Scholar] [CrossRef]
- Mody, R.J.; Prensner, J.R.; Everett, J.; Parsons, D.W.; Chinnaiyan, A.M. Precision medicine in pediatric oncology: Lessons learned and next steps. Pediatr. Blood Cancer 2017, 64, e26288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilarczyk, M.; Fazel-Najafabadi, M.; Kouril, M.; Shamsaei, B.; Vasiliauskas, J.; Niu, W.; Mahi, N.; Zhang, L.; Clark, N.A.; Ren, Y.; et al. Connecting omics signatures and revealing biological mechanisms with iLINCS. Nat. Commun. 2022, 13, 4678. [Google Scholar] [CrossRef] [PubMed]
- Serrano Lopez, J.; Jimenez-Jimenez, C.; Chutipongtanate, S.; Serrano, J.; Rodriguez-Moreno, M.; Jimenez, A.; Jimenez, Y.; Pedrero, S.G.; Lainez, D.; Alonso-Dominguez, J.M.; et al. High-throughput RNA sequencing transcriptome analysis of ABC-DLBCL reveals several tumor evasion strategies. Leuk. Lymphoma 2022, 63, 1861–1870. [Google Scholar] [CrossRef]
- Venkatraman, S.; Balasubramanian, B.; Pongchaikul, P.; Tohtong, R.; Chutipongtanate, S. Molecularly Guided Drug Repurposing for Cholangiocarcinoma: An Integrative Bioinformatic Approach. Genes 2022, 13, 271. [Google Scholar] [CrossRef]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Cripe, T.P. Immunotherapies for Pediatric Solid Tumors: A Targeted Update. Paediatr. Drugs 2022, 24, 1–12. [Google Scholar] [CrossRef]
- Dhillon, S. Dinutuximab: First global approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Doussau, A.; Geoerger, B.; Jimenez, I.; Paoletti, X. Innovations for phase I dose-finding designs in pediatric oncology clinical trials. Contemp Clin. Trials 2016, 47, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Marshall, L.V.; Binner, D.; Herold, R.; Rousseau, R.; Blanc, P.; Capdeville, R.; Carleer, J.; Copland, C.; Kerloeguen, Y.; et al. Joint adolescent-adult early phase clinical trials to improve access to new drugs for adolescents with cancer: Proposals from the multi-stakeholder platform-ACCELERATE. Ann. Oncol. 2018, 29, 766–771. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Friedrich, P.; Alcasabas, P.; Antillon, F.; Banavali, S.; Castillo, L.; Israels, T.; Jeha, S.; Harif, M.; Sullivan, M.J.; et al. Toward the Cure of All Children with Cancer Through Collaborative Efforts: Pediatric Oncology As a Global Challenge. J. Clin. Oncol. 2015, 33, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.; Walsh, J.A.; Weinrich, S.L.; Beaupre, D.; Blasi, E.; Arenson, D.R.; Jacobs, I.A. Navigating the Regulatory Landscape to Develop Pediatric Oncology Drugs: Expert Opinion Recommendations. Paediatr. Drugs 2021, 23, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Bender, J.G.; Macy, M.E.; Moreno, L. Opportunities and Challenges in Drug Development for Pediatric Cancers. Cancer Discov. 2021, 11, 545–559. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cancer Predisposition Syndrome | Common Solid Tumors | Mutated Genes (Inheritance) | Dysregulated Pathways | Reference |
|---|---|---|---|---|
| Beckwith–Wiedemann syndrome | Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma | CDKN1C (AD) | Cell cycle | [39,40] |
| Constitutional mismatch repair deficiency | Brain tumor, neuroblastoma, Wilms tumor, osteosarcoma, rhabdomyosarcoma | MLH1, MSH2, MSH6, PMS2 (AR) | DNA mismatch repair | [36,41] |
| Hereditary retinoblastoma | Retinoblastoma, melanoma, osteosarcoma, pineoblastoma | RB1 (AD) | Cell cycle | [39,42] |
| Li-Fraumeni syndrome | Brain tumor, sarcoma, neuroblastoma, rhabdomyosarcoma, retinoblastoma | TP53 (AD) | Cell cycle, apoptosis | [39,43,44] |
| Neurofibromatosis | Glioma, astrocytoma, ependymoma, malignant peripheral nerve sheath tumors, neuroblastoma, rhabdomyosarcoma | NF1, NF2 (AD) | RAS/MAPK | [39,45] |
| Rhabdoid tumor predisposition syndrome | Atypical teratoid/rhabdoid tumor, malignant rhabdoid tumor | SMARCB1, SMARCA4 (AD) | Wnt/β-catenin, Sonic hedgehog | [39,46] |
| Multiple endocrine neoplasia | Ependymoma, Medullary thyroid cancer | MEN1, RET (AD) | Transcriptional activity | [39,47] |
| Nevoid basal cell carcinoma | Medulloblastoma, rhabdomyosarcoma | PTCH1, PTCH2, SUFU (AD) | Sonic hedgehog | [39,46] |
| Familial adenomatous polyposis | Medulloblastoma, hepatoblastoma | APC (AD) | Wnt/β-catenin | [39,48] |
| Tuberous sclerosis | Subependymal giant cell astrocytoma, rhabdomyosarcoma | TSC1, TSC2 (AD) | mTOR | [39,49] |
| Bloom syndrome | Osteosarcoma, Wilms tumor | BLM (AR) | DNA double-strand repair | [34,35] |
| Rubinstein–Taybi syndrome | Medulloblastoma, neuroblastoma, rhabdomyosarcoma | CREBBP (AD) | Transcriptional regulation | [34,35] |
| Noonan syndrome | Rhabdomyosarcoma, neuroblastoma, glioma, hepatoblastoma | PTPN11, SOS1, RAF1, KRAS, MAP2K1 (AD) | RAS/MAPK | [50] |
| Gene | Alterations | Targeted Therapies | Cancer Types | FDA-Approved Level a |
|---|---|---|---|---|
| AKT1 | E17K | AZD5363 | Breast Cancer, Ovarian Cancer; Endometrial Cancer | Lv.3 |
| ALK | Fusions | Alectinib; Brigatinib; Ceritinib; Crizotinib | Non-Small Cell Lung Cancer | Lv.1 |
| Brigatinib; Ceritinib; Crizotinib | Inflammatory Myofibroblastic Tumor | Lv.2 | ||
| Oncogenic Mutations | Lorlatinib | Non-Small Cell Lung Cancer; Neuroblastoma c | Lv.1 | |
| Crizotinib | Non-Small Cell Lung Cancer; Neuroblastoma c | Lv.R2 | ||
| ARAF | Oncogenic Mutations | Sorafenib | Non-Small Cell Lung Cancer | Lv.3 |
| ARID1A | Truncating Mutations | PLX2853; Tazemetostat | All Solid Tumors | Lv.4 |
| ATM | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| BRAF | V600E | Dabrafenib + Trametinib | Melanoma; Non-Small Cell Lung Cancer; Low grade glioma b; High grade glioma b | Lv.1 |
| Encorafenib + Cetuximab | Colorectal Cancer | |||
| Fusions or V600E | Selumetinib | Pilocytic Astrocytoma | Lv.2 | |
| V600E | Dabrafenib + Trametinib, Vemurafenib + Cobimetinib | Diffuse Glioma; Encapsulated Glioma; Ganglioglioma | ||
| Fusions | Trametinib; Cobimetinib | Ovarian Cancer | Lv.3 | |
| V600E | Dabrafenib + Trametinib | Biliary Tract Cancer | ||
| G464, G469A, G469R, G469V, K601, L597 | PLX8394 | All Solid Tumors | Lv.4 | |
| BRCA1/2 | Oncogenic Mutations | Niraparib; Olaparib; Olaparib + Bevacizumab; Rucaparib | Ovarian Cancer; Peritoneal Serous Carcinoma | Lv.1 |
| Olaparib; Rucaparib | Prostate Cancer | |||
| Olaparib; Talazoparib | Breast Cancer | Lv.3 | ||
| BRIP1 | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| CDK4 | Amplification | Palbociclib; Abemaciclib | Dedifferentiated Liposarcoma; Well-Differentiated Liposarcoma | Lv.4 |
| CDK12 | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| CDKN2A | Oncogenic Mutations | Palbociclib; Ribociclib; Abemaciclib | All Solid Tumors | Lv.4 |
| CHEK1/2 | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| EGFR | Exon 19 deletion, L858R | Afatinib; Dacomitinib; Erlotinib; Erlotinib + Ramucirumab; Gefitinib; Osimertinib | Non-Small Cell Lung Cancer | Lv.1 |
| Exon 20 insertion | Amivantamab; Mobocertinib | |||
| G719, L861Q, S768I | Afatinib | |||
| T790M | Osimertinib | |||
| A763_Y764insFQEA | Erlotinib | Lv.2 | ||
| E709_T710delinsD | Afatinib | Lv.3 | ||
| Exon 19 insertion | Erlotinib; Gefitinib | |||
| Exon 20 insertion | Poziotinib | |||
| Kinase Domain Duplication | Afatinib | |||
| A763_Y764insFQEA or Exon 19 insertion or L718V, L747P | Afatinib | Lv.4 | ||
| D761Y | Osimertinib | |||
| Kinase Domain Duplication | Erlotinib; Gefitinib | |||
| Amplification or A289V, R108K, T263P | Lapatinib | Glioma | ||
| Exon 20 insertion, T790M | Erlotinib; Gefitinib; Afatinib | Non-Small Cell Lung Cancer | Lv.R1 | |
| C797S, D761Y, G724S, L718V | Osimertinib; Gefitinib | Lv.R2 | ||
| ERBB2 | Amplification | Ado-Trastuzumab; Emtansine; Lapatinib + Capecitabine; Lapatinib + Letrozole, Margetuximab + Chemotherapy; Neratinib; Neratinib + Capecitabine; Trastuzumab + Pertuzumab + Chemotherapy; Trastuzumab + Tucatinib + Capecitabine; Trastuzumab Deruxtecan; Trastuzumab, Trastuzumab + Chemotherapy | Breast Cancer | Lv.1 |
| Pembrolizumab + Trastuzumab + Chemotherapy; Trastuzumab + Chemotherapy; Trastuzumab Deruxtecan | Esophagogastric Cancer | Lv.1 | ||
| Trastuzumab + Lapatinib; Trastuzumab + Pertuzumab; Trastuzumab Deruxtecan | Colorectal Cancer | Lv.2 | ||
| Oncogenic Mutations | Ado-Trastuzumab; Emtansine; Trastuzumab Deruxtecan | Non-Small Cell Lung Cancer | Lv.2 | |
| Neratinib | Breast Cancer; Non-Small Cell Lung Cancer | Lv.3 | ||
| ESR1 | Oncogenic Mutations | AZD9496; Fulvestrant | Breast Cancer | Lv.3 |
| FANCL | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| FGFR1 | Amplification | Debio1347; Infigratinib; Erdafitinib | Lung Squamous Cell Carcinoma | Lv.3 |
| Oncogenic Mutations | Debio1347; Infigratinib; Erdafitinib; AZD4547 | All Solid Tumors | Lv.4 | |
| FGFR2 | Fusions | Erdafitinib | Bladder Cancer | Lv.1 |
| Infigratinib; Pemigatinib | Cholangiocarcinoma | |||
| Oncogenic Mutations | Debio1347; Infigratinib; Erdafitinib; AZD4547 | All Solid Tumors | Lv.4 | |
| FGFR3 | Fusions or G370C, R248C, S249C, Y373C | Erdafitinib | Bladder Cancer | Lv.1 |
| G380R, K650, S371C | Erdafitinib | Lv.3 | ||
| Oncogenic Mutations | Debio1347; Infigratinib; Erdafitinib; AZD4547 | All Solid Tumors | Lv.4 | |
| FLI1 | EWSR1-FLI1 Fusion | TK216 | Ewing Sarcoma | Lv.4 |
| HRAS | Oncogenic Mutations | Tipifarnib | Bladder Urothelial Carcinoma; Head and Neck Squamous Cell Carcinoma | Lv.3 |
| IDH1 | R132 | Ivosidenib | Cholangiocarcinoma | Lv.1 |
| Oncogenic Mutations | Chondrosarcoma | Lv.2 | ||
| R132 | Glioma | Lv.3 | ||
| KDM6A | Oncogenic Mutations | Tazemetostat | Bladder Cancer | Lv.4 |
| KIT | A502_Y503dup, K509I, N505I, S476I, S501_A502dup, A829P and 5 other alterations, D572A and 65 other alterations, K642E, T670I, V654A | Imatinib; Regorafenib; Ripretinib; Sunitinib | Gastrointestinal Stromal Tumor | Lv.1 |
| A829P and 5 other alterations | Sorafenib | Gastrointestinal Stromal Tumor | Lv.2 | |
| KRAS | G12C | Sotorasib | Non-Small Cell Lung Cancer | Lv.1 |
| Adagrasib | Non-Small Cell Lung Cancer | Lv.3 | ||
| Adagrasib; Adagrasib + Cetuximab | Colorectal Cancer | |||
| Oncogenic Mutations | Cobimetinib; Trametinib; Binimetinib | All Solid Tumors | Lv.4 | |
| MAP2K1 | Oncogenic Mutations | Cobimetinib; Trametinib | Melanoma; Non-Small Cell Lung Cancer; Low grade glioma c | Lv.3 |
| MDM2 | Amplification | Milademetan | Dedifferentiated Liposarcoma; Well-Differentiated Liposarcoma | Lv.4 |
| MET | D1010, Exon 14 deletion, Exon 14 splice mutation | Capmatinib; Tepotinib | Non-Small Cell Lung Cancer | Lv.1 |
| Amplification or D1010, Exon 14 deletion, Exon 14 splice mutation | Crizotinib | Lv.2 | ||
| Y1003mut | Tepotinib; Capmatinib; Crizotinib | Lv.3 | ||
| Fusions | Crizotinib | All Solid Tumors | Lv.4 | |
| MTOR | E2014K, E2419K | Everolimus | Bladder Cancer | Lv.3 |
| Q2223K | Everolimus | Renal Cell Carcinoma | ||
| L2209V, L2427Q | Temsirolimus | |||
| Oncogenic Mutations | Everolimus; Temsirolimus | All Solid Tumors, Rhabdomyosarcoma c | Lv.4 | |
| NF1 | Oncogenic Mutations | Selumetinib | Neurofibroma b | Lv.1 |
| Trametinib; Cobimetinib | All Solid Tumors | Lv.4 | ||
| NRG1 | Fusions | Zenocutuzumab | All Solid Tumors | Lv.3 |
| NTRK1/2/3 | Fusions | Entrectinib; Larotrectinib | All Solid Tumors b | Lv.1 |
| PALB2 | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| PDGFB | COL1A1-PDGFB Fusion | Imatinib | Dermatofibrosarcoma Protuberans | Lv.1 |
| PDGFRA | Exon 18 in-frame deletions or insertions, Exon 18 missense mutations | Avapritinib | Gastrointestinal Stromal Tumor | Lv.1 |
| Oncogenic Mutations | Regorafenib | Gastrointestinal Stromal Tumor; Medullary thyroid cancer c, Hepatocellular carcinomac | Lv.2 | |
| Imatinib; Ripretinib; Sunitinib | Gastrointestinal Stromal Tumor | |||
| D842V | Dasatinib | |||
| D842V | Imatinib | Gastrointestinal Stromal Tumor | Lv.R1 | |
| PIK3CA | C420R and 10 other alterations | Alpelisib + Fulvestrant | Breast Cancer | Lv.1 |
| Oncogenic Mutations (excluding C420R, E542K, E545A, E545D, E545G, E545K, Q546E, Q546R, H1047L, H1047R and H1047Y) | Alpelisib + Fulvestrant | Lv.2 | ||
| PTCH1 | Truncating Mutations | Sonidegib; Vismodegib | Medulloblastoma | Lv.3 |
| PTEN | Oncogenic Mutations | GSK2636771; AZD8186 | All Solid Tumors | Lv.4 |
| RAD51B, RAD51C, RAD51D, RAD54L | Oncogenic Mutations | Olaparib | Prostate Cancer | Lv.1 |
| RET | Fusions or Oncogenic Mutations | Pralsetinib; Selpercatinib | Non-Small Cell Lung Cancer, Thyroid Cancer, Medullary Thyroid Cancer b | Lv.1 |
| Fusions | Cabozantinib | Non-Small Cell Lung Cancer; Sarcoma c | Lv.2 | |
| Vandetanib | Non-Small Cell Lung Cancer | Lv.3 | ||
| ROS1 | Fusions | Crizotinib | Non-Small Cell Lung Cancer | Lv.1 |
| Entrectinib | Biomarker (+), solid and brain b | |||
| SMARCB1 | Deletion | Tazemetostat | Epithelioid Sarcoma | Lv.1 |
| STK11 | Oncogenic Mutations | Bemcentinib + Pembrolizumab | Non-Small Cell Lung Cancer | Lv.4 |
| TSC1/2 | Oncogenic Mutations | Everolimus | Encapsulated Glioma; Subependymal giant cell astrocytoma b | Lv.1 |
| Tumor | Significantly Mutated Genes (# Prevalence) |
|---|---|
| Medulloblastoma | DDX3X (5.8%), KMT2D (5.8%), CTNNB1 (5.5%), PTCH1 (5.1%), TP53 (4.0%), SMARCA4 (3.6%), KDM6A (3.1%), SUFU (1.3%), SMO (1.5%), KMT2C (1.4%), CREBBP (1.3%), APC † (0.6%), IDH1 (0.4%) |
| High grade glioma | TP53†‡(28.5%), ATRX (11.3%), PIK3CA (5.6%), PDGFRA‡(5.1%), BCOR (3.0%), PPM1D‡(3.9%), CREBBP‡(1.8%), NF1†(0.8%), EGFR‡(0.6%) |
| Ependymoma | RELA‡(25.0%), IGF2R†(20.0%) |
| Low grade glioma | FGFR1‡(33.3%), BRAF (8.7%), NF1†(3.9%), KIAA1549 (1.9%) |
| Neuroblastoma | MYCN (36.2%), MYCNOS (33.0%), ATRX (22.2%), DDX1 (22.3%), ALK (1.4%), RYR1 (0.5%), PTPN11 (0.7%) |
| Wilms tumor | MYCN (12.4%), MYCNOS (12.4%), TP53 (3.2%), DROSHA‡(1.8%), WT1 (1.6%), CTNNB1 (1.5%), DGCR8 (1.1%) |
| Osteosarcoma | TP53†(30.0%), RB1†(15.4%), ATRX (9.7%) |
| Ewing’s sarcoma | EWSR1 (29.6%), FLI1 (25.9%), ERG (4.7%), STAG2 (2.4%) |
| Retinoblastoma | RB1†(51.6%), BCOR (3.2%) |
| Rhabdomyosarcoma | PAX3‡(28.6%), FOXO1‡(25.9%), PAX7‡(16.7%), TP53†‡(12.3%), FGFR4‡(7.7%), NRAS‡(4.6%) |
| Signaling Pathway | Gene | Alterations | Effected Domain | Pediatric CANCER Types | Potentially Targeted Therapy (Level of Evidence) | Additional References for Targeted Therapy |
|---|---|---|---|---|---|---|
| Tyrosine Kinase | ALK | Fusion | NBL | Crizotinib, Ceritinib, Alectinib, Lorlatinib | cBioPortal | |
| F1174L ‡ | CAD exon23 | NBL | Crizotinib (B) | [74,75] | ||
| F1245V | CAD exon24 | NBL | ||||
| R1275Q/L †‡ | CAD exon25 | NBL | ||||
| NTRK1 | TPM3::NTRK1 | HGG | Larotrectinib (A) | [18,76,77] | ||
| NTRK2 | Fusion | HGG, LGG | Larotrectinib (A) | [77,78,79] | ||
| NTRK3 | ETV6::NTRK3 | HGG, LGG | Larotrectinib (A) | [76,77] | ||
| PDGFRA | Y288C | Exon6 | HGG | Imatinib, sunitinib, regorafenib and ripretinib | cBioPortal | |
| E311_E7splice | Exon7 | HGG | ||||
| N659K ‡ | PKD exon14 | HGG | Imatinib, sunitinib, regorafenib and ripretinib | cBioPortal | ||
| D842Y | PKD exon18 | HGG | Avapritinib, Imatinib, Sunitinib | cBioPortal | ||
| ROS1 | Fusion | OS, HGG | Crizotinib, Entrectinib | cBioPortal | ||
| MAPK signaling | NF1 | Fusion | OS, NBL, MB, HGG | Trametinib, Cobimetinib | cBioPortal | |
| Mutation | LGG, NBL | Selumetinib (B) | [80,81,82] | |||
| BRAF | KIAA1549::BRAF | LGG, PA | Selumetinib (B), Sorafenib (C) | [81,83,84] | ||
| V600E | LGG, HGG, PA, NBL | Selumetinib (B), Vemurafenib (B), Dabrafenib (B) | [81,85,86] | |||
| KRAS | G12D | GTPase exon2 | LGG, NBL | Trametinib, Cobimetinib, Binimetinib | cBioPortal | |
| NRAS | G12S | GTPase exon2 | HGG | Binimetinib, Binimetinib + Ribociclib | cBioPortal | |
| Q61K ‡/R | GTPase exon3 | RHB, NBL | ||||
| PTPN11 | E69K | Exon3 | NBL, PA | |||
| A72T/D | Exon3 | NBL | ||||
| E76A | Exon3 | NBL, PA | ||||
| Notch signaling | NOTCH2 | Fusion | OS, NBL | |||
| R5_P6fs | Exon1 | OS, NBL, RHB | ||||
| P6fs | Exon1 | NBL, MB, PA, WLM | ||||
| Sonic hedgehog signaling | PTCH1 | Mutation | MB | Sonidegib (B) | [87] | |
| A300fs | Exon6 | MB | Sonidegib, Vismodegib | cBioPortal | ||
| Y804fs | Exon15 | MB | Sonidegib, Vismodegib | cBioPortal | ||
| SMO | L412F | MB | Vismodegib # (C) | [88] | ||
| W535L | MB | Vismodegib # | cBioPortal | |||
| Wnt signaling | CTNNB1 | D32 | Exon3 | MB | ||
| S33 | Exon3 | MB | ||||
| G34 | Exon3 | MB, RHB, ACT, HB | ||||
| S37 | Exon3 | MB | ||||
| T41A/N | Exon3 | WLM, MB, RHB | ||||
| N387K ‡ | Exon8 | WLM | ||||
| PI3K signaling | PTEN | Fusion | OS | GSK2636771, AZD8186 | cBioPortal | |
| R130 | CAD exon5 | HGG | ||||
| R233 * | Exon7 | HGG | ||||
| PIK3CA | R88Q | SBD exon2 | HGG | Alpelisib + Fulvestrant | cBioPortal | |
| N345K ‡ | Exon5 | MB, RHB, EPD | ||||
| E545K | Exon10 | HGG | ||||
| Q546K | Exon10 | HGG, MB | ||||
| E888 * | CAD exon18 | NBL | ||||
| H1047R/L | CAD exon21 | HGG, MB, RHB, NBL | ||||
| FGFR1 | Fusion | LGG | Erdafitinib, Infigratinib | cBioPortal | ||
| Internal tandem duplication | CAD | LGG | ||||
| N546K | CAD exon12 | LGG, NBL, PA, WLM, HGG | Pemigatinib (C) | [89] | ||
| K656E | CAD exon14 | PA, HGG, WLM | Erdafitinib, Infigratinib | cBioPortal | ||
| FGFR4 | V550L ‡ | CAD exon13 | RHB | |||
| EGFR | A289V | Exon7 | HGG | Lapatinib | cBioPortal | |
| TGFB signaling | ACVR1 | R206H | CAD exon6 | HGG | ||
| R258G | CAD exon7 | HGG | ||||
| G328E/V | CAD exon8 | HGG | ||||
| G356_E9splice | CAD exon9 | HGG | ||||
| Cell cycle and DNA repair | RB1 | Fusion | OS | |||
| W78 * | Exon2 | OS | ||||
| R320 * | Exon10 | RB, HGG | ||||
| R445 *† | Exon14 | RB | ||||
| R552 * | Exon17 | RB, OS, HGG | ||||
| R579 * | Exon18 | RB | ||||
| TP53 | Mutation | HGG, WLM, OS, MB | Vismodegib (C) | [90] | ||
| T125T/R † | DBD exon4 | HGG, WLM, ACT | ||||
| R175H †‡ | DBD exon5 | HGG, WLM, MB, RHB, ACT | ||||
| C176F | DBD exon5 | RHB, EWS, NBL | ||||
| R213 *† | DBD exon6 | HGG, MB | ||||
| G245S | DBD exon7 | HGG, MB | ||||
| R248Q/W † | DBD exon7 | MB, HGG, OS, WLM | ||||
| R273C †/H | DBD exon4 | HGG, EWS, ACT, MB, OS | ||||
| R282W † | DBD exon8 | OS, HGG, MB | ||||
| R337H † | Exon10 | ACT | ||||
| R342 */P | Exon10 | HGG, WLM | ||||
| CDK1 | V124G | CAD exon5 | MB | |||
| PPM1D | W427 * | Exon6 | HGG | |||
| S516 * | Exon6 | HGG, NBL | ||||
| E525 * | Exon6 | HGG, MB | ||||
| Transcriptional regulation | EWSR1 | FLI1::EWSR1 | EWS | TK216 | cBioPortal | |
| ERG::EWSR1 | EWS | |||||
| BCOR | R1164* | Exon7 | HGG | |||
| H1481fs | Exon11 | HGG | ||||
| SIX1 | Q177R | DBD exon1 | WLM | |||
| MYCN | Fusion | NBL | ||||
| P44L | Exon2 | WLM, NBL, MB | ||||
| PAX7 | FOXO1::PAX7 | RHB | ||||
| PAX3 | FOXO1::PAX3 | RHB | ||||
| RNA processing | DROSHA | E1147K | Ribonuclease exon29 | WLM | ||
| D1151 | Ribonuclease exon29 | WLM, NBL | ||||
| DGCR8 | E518K | RBM exon7 | WLM | |||
| DDX1 | DDX1::DDX1 | NBL | ||||
| MYCN::DDX1 | NBL | |||||
| DDX3X | R351W | HD exon11 | MB | |||
| M380I | HD exon11 | MB | ||||
| R534 | HD exon14 | MB | ||||
| Epigenetics | ATRX | ATRX::ATRX | NBL | |||
| N294fs | Exon9 | OS | ||||
| ASXL1 | R643fs | Exon13 | WLM | |||
| R693 * | Exon13 | HGG, EPD | ||||
| H3-3A (H3F3A) | K28M | Exon2 | HGG, LGG | |||
| G35R | Exon2 | HGG | ||||
| KMT2C | T1636P | Exon33 | MB | |||
| E2798fs | Exon38 | MB | ||||
| I4084L | Exon48 | MB | ||||
| SMARCA4 | T910M | HD exon19 | MB | |||
| H3C2 (HIST1H3B) | K28M ‡ | Exon1 | HGG | |||
| KDM6A | S54_E2splice | Exon2 | MB | |||
| R1351 * | Exon28 | MB | ||||
| IDH1 | R132C/H | Exon4 | MB, HGG, LGG | Bevacizumab and Sunitinib (B) | [91] | |
| R222C/H | Exon6 | HGG, EWS | ||||
| RELA | Fusion | EPD, HGG | ||||
| STAG2 | R216 * | STAG domain exon8 | EWS | |||
| R259 * | STAG domain exon9 | MB, HGG | ||||
| E1209Q | Exon33 | OS | ||||
| FLI1 | EWSR1::FLI1 | EWS | ||||
| ERG | EWSR1::ERG | EWS |
| Gene Involved in Trial Design | NCT (Recruitment Status) | Phase | Specification | Intervention(s) | Cancer Type(s) | Eligibility | Enrollment (Number) |
|---|---|---|---|---|---|---|---|
| ALK | NCT01742286 (D) | I | ALK alterations | Ceritinib | ALK-activated Tumors | 1–17 years | 83 |
| NCT02465528 (C) | II | ALK alterations | Ceritinib | Tumors With Aberrations in ALK, Glioblastoma | ≥18 years | 22 | |
| NCT02780128 (A) | I | ALK mutation | Ceritinib + Ribociclib | Neuroblastoma | 1–21 years | 131 | |
| NCT03107988 (A) | I | ALK alterations | Lorlatinib + Chemotherapy | Neuroblastoma | ≥1 year | 65 | |
| NCT03194893 (B) | III | ALK alterations | Alectinib or Crizotinib | Neoplasms | all | 200 | |
| NCT04774718 (A) | I, II | ALK fusion | Alectinib | ALK Fusion-positive Solid or CNS Tumors | ≤17 years | 42 | |
| NCT05384626 (A) | I, II | ALK alterations | NVL-655 | Solid Tumor, NSCLC | ≥12 years | 214 | |
| BRAF | NCT01089101 (B) | I, II | BRAF V600E mutation or BRAF-KIAA1549 fusion | Selumetinib | Low Grade Glioma, Recurrent Childhood Pilocytic Astrocytoma, Recurrent Neurofibromatosis Type 1 | 3–21 years | 220 |
| NCT01596140 (D) | I | BRAF mutation | Vemurafenib + Everolimus or Temsirolimus | Advanced Cancer, Solid Tumor | all | 27 | |
| NCT01636622 (D) | I | BRAF mutation | Vemurafenib + Chemotherapy | Advanced Cancers | ≥12 years | 21 | |
| NCT01677741 (D) | I, II | BRAF V600 mutation | Dabrafenib | Neoplasms, Brain | 1–17 years | 85 | |
| NCT02124772 (D) | I, II | BRAF V600 mutation | Dabrafenib + Trametinib | Solid Tumors, neuroblastoma, low grade glioma, neurofibromatosis Type 1 | 1 month to 17 years | 139 | |
| NCT02684058 (B) | II | BRAF V600 mutation | Dabrafenib + Trametinib + Radiation | Solid Tumors, CNS Tumors, high grade glioma, low grade glioma | 1–17 years | 149 | |
| NCT03919071 (A) | II | BRAF V600 mutation | Dabrafenib + Trametinib + Radiation | Anaplastic Astrocytoma, Glioblastoma, Malignant Glioma | 1–21 years | 58 | |
| NCT04576117 (A) | III | BRAF rearrangement | Selumetinib + Chemotherapy | Low Grade Astrocytoma, Glioma | 2–25 years | 18 | |
| EGFR | NCT00198159 (C) | II | EGFR expression | Gefitinib | Refractory Germ Cell Tumors Expressing EGRF | ≥15 years | 21 |
| NCT00418327 (D) | I | EGFR mutation | Erlotinib + Radiation | Malignant Brain Tumor, Glioma | 1–21 years | 48 | |
| NCT01182350 (C) | II | EGFR overexpression | Erlotinib + Bevacizumab + Temozolomide + Radiation | Diffuse Intrinsic Pontine Glioma | 3–18 years | 53 | |
| NCT01962896 (C) | II | EGFR/mTOR pathway activation | Erlotinib + Sirolimus | Relapsed/Recurrent Germ Cell Tumors | 1–50 years | 4 | |
| EWSR1 | NCT03709680 (A) | II | EWSR1-ETS or FUS-ETS rearrangement | Palbociclib + Chemotherapy | Ewing Sarcoma, Rhabdomyosarcoma, Neuroblastoma, Medulloblastoma, Diffuse Intrinsic Pontine Glioma | 2–20 years | 184 |
| NCT04129151 (B) | II | EWSR1 or FUS translocation | Palbociclib + Ganitumab | Ewing Sarcoma | 12–50 years | 18 | |
| FGFR | NCT04083976 (A) | II | FGFR alteration | Erdafitinib | Advanced Solid Tumor | ≥6 years | 336 |
| NCT05180825 (A) | II | FGFR1 and MYB/MYBL1 alterations, 7q34 duplication | Trametinib or Vinblastine | Grade 1 Glioma, Mixed Glio-neuronal Tumors, Pleomorphic Xanthoastrocytoma | 1 month to 25 years | 134 | |
| H3 | NCT02525692 (B) | II | H3 K27M mutation | ONC201 | Glioblastoma, Glioma | ≥16 years | 89 |
| NCT03416530 (A) | I | H3 K27M mutation | ONC201 | Diffuse Intrinsic Pontine Glioma, Glioma, Malignant | 2–18 years | 130 | |
| NCT05009992 (A) | II | H3 K27M mutation | ONC201 + Paxalisib or Radiation | Diffuse Intrinsic Pontine Glioma, Diffuse Midline Glioma, H3 K27M-Mutant | 2–39 years | 216 | |
| IDH | NCT03749187 (A) | I | IDH1/2 mutation | PARP Inhibitor BGB-290 + Chemotherapy | Glioblastoma, Glioma | 13–39 years | 78 |
| MYCN | NCT02559778 (A) | II | MYCN amplification | Ceritinib, Dasatinib, Sorafenib or Vorinostat + Chemotherapy | Neuroblastoma | ≤22 years | 500 |
| NCT03126916 (A) | III | MYCN amplification | Lorlatinib + Standard therapy | Ganglioneuroblastoma, Neuroblastoma | 1–30 years | 658 | |
| NF | NCT01158651 (D) | II | NF1 mutation | Everolimus | Glioma | 1–21 years | 23 |
| NCT03095248 (A) | II | NF2 mutation | Selumetinib | Neurofibromatosis 2, Vestibular Schwannoma, Meningioma, Ependymoma, Glioma | 3–45 years | 34 | |
| NCT03326388 (A) | I, II | NF1 positive | Selumetinib | Neurofibromatosis Type 1, Plexiform Neurofibroma, Optic Nerve Glioma | 3–18 years | 30 | |
| NCT03871257 (A) | III | NF1 positive | Selumetinib + Chemotherapy | Low Grade Glioma, Neurofibromatosis Type 1, Visual Pathway Glioma | 2–21 years | 290 | |
| NTRK | NCT02637687 (A) | I, II | NTRK-fusion | Larotrectinib | Solid Tumors Harboring NTRK Fusion | ≤21 years | 155 |
| NCT03834961 (A) | II | NTRK-fusion | Larotrectinib | Solid Tumor, CNS Tumor | ≤30 years | 70 | |
| NCT04879121 (A) | II | NTRK amplification | Larotrectinib | Solid Neoplasm | ≥16 years | 13 | |
| PDGFR | NCT00417807 (D) | I, II | PDGFR expression | Imatinib | Refractory Desmoplastic Small Round Cell Tumors | ≥16 years | 9 |
| NCT03352427 (C) | II | PDGFR alteration | Dasatinib + Everolimus | Glioma, High Grade Glioma, Pontine Tumors | 1–50 years | 3 | |
| Rb1 | NCT02255461 (C) | I | Rb1 positive | Palbociclib | CNS Tumors, Solid Tumors | 4–21 years | 35 |
| NCT03355794 (B) | I | Rb1 positive | Everolimus + Ribociclib | Diffuse Intrinsic Pontine Glioma, Malignant Glioma of Brain, High Grade Glioma, Glioblastoma, Anaplastic Astrocytoma | 1–30 years | 24 | |
| NCT03387020 (D) | I | Rb1 positive | Everolimus + Ribociclib | CNS Tumors | 1–21 years | 22 | |
| ALK c-MET ROS | NCT00939770 (D) | I, II | ALK or MET alterations | Crizotinib | Recurrent Neuroblastoma | 1–21 years | 122 |
| NCT01524926 (B) | II | ALK or MET pathway activation | Crizotinib | Lymphoma, Sarcoma, Rhabdomyosarcoma | ≥1 year | 582 | |
| NCT02034981 (B) | II | ALK, MET or ROS1 alterations | Crizotinib | Solid Tumors | ≥1 year | 246 | |
| NCT02650401 (A) | I, II | ALK, ROS1, or NTRK1-3 Rearrangements | Entrectinib | Solid Tumors, CNS Tumors, Neuroblastoma | ≤18 years | 68 | |
| NCT03093116 (A) | I, II | ALK, ROS1, or NTRK1-3 Rearrangements | Repotrectinib | Solid tumor, CNS tumor | ≥12 years | 500 | |
| RAS RAF MEK ERK NF1 | NCT02285439 (B) | I, II | BRAF truncated fusion or NF1 mutation | MEK162 | Low-Grade Gliomas, Brain, Soft Tissue Neoplasms | 1–18 years | 105 |
| NCT02639546 (D) | I, II | RAS/RAF/MEK/ERK pathway activation | Cobimetinib | Solid Tumors | 6 months to 30 years | 56 | |
| NCT03363217 (A) | II | BRAF-KIAA1549 fusion, NF1 mutation, MAPK/ERK pathway activation | Trametinib | Low-grade Glioma, Plexiform Neurofibroma, Central Nervous System Glioma | 1 month to 25 years | 150 | |
| NCT04201457 (A) | I, II | BRAF V600 mutation or truncated fusion, NF1 mutation | Dabrafenib + Trametinib + hydroxychloroquine | Low Grade Glioma, High Grade Glioma | 1–30 years | 75 | |
| NCT04216953 (A) | I, II | MAPK pathway status and Tumor Mutational Burden | Cobimetinib + Atezolizumab | Sarcoma, Soft Tissue | ≥6 months | 120 | |
| SHH WNT | NCT00822458 (D) | I | SHH or WNT signaling activation | Vismodegib | Recurrent Childhood Medulloblastoma | 3–21 years | 34 |
| NCT01239316 (D) | II | SHH signaling activation | Vismodegib | Recurrent Childhood Medulloblastoma | 3–21 years | 12 | |
| NCT01878617 (A) | II | SHH or WNT signaling activation | Vismodegib + chemotherapy | Medulloblastoma | 3–39 years | 660 | |
| Others | NCT01396408 (B) | II | Mutations in sunitinib targets such as VEGFR, PDGFR, KIT, RET or mutations in mTOR pathway such as PTEN, TS1/2, LKB1, NF1/2 | Sunitinib or temsirolimus | Advanced Rare Tumors | ≥16 years | 137 |
| NCT03654716 (A) | I | MDM2, MDMX, PPM1D or TET2 amplification | ALRN-6924 | Solid Tumor, CNS Tumor | 1–21 years | 69 |
| Gene Involved in Trial Design | NCT (Recruitment Status) | Phase | Specification | Intervention(s) | Cancer Type(s) | Eligibility | Enrollment (Number) |
|---|---|---|---|---|---|---|---|
| Testing the Use of Food and Drug Administration (FDA)-Approved Drugs (TAPUR) | NCT02693535 (A) | II | ALK, ROS1, MET | Crizotinib | Advanced Solid Tumors | ≥12 years | 3581 |
| CDKN2A, CDK4, CDK6 | Palbociclib or Abemaciclib | ||||||
| CSF1R, PDGFR, VEGFR | Sunitinib | ||||||
| mTOR, TSC | Temsirolimus | ||||||
| BRAF V600E/D/K/R | Vemurafenib and Cobimetinib | ||||||
| RET, VEGFR1/2/3, KIT, PDGFRβ, RAF-1, BRAF | Regorafenib | ||||||
| BRCA1/2, ATM | Olaparib | ||||||
| NRG1 | Afatinib | ||||||
| BRCA1/2, PALB2 | Talazoparib | ||||||
| ROS1 fusion | Entrectinib | ||||||
| NTRK amplification | Larotrectinib | ||||||
| NCI-COG Pediatric MATCH Screening | NCT03155620 (A) | II | NTRK1, NTRK2, or NTRK3 gene fusion | Larotrectinib | Refractory or Recurrent Advanced Solid Tumors | 1–21 years | 2316 |
| FGFR1, FGFR2, FGFR3, or FGFR4 gene mutation | Erdafitinib | ||||||
| EZH2, SMARCB1, or SMARCA4 gene mutation | Tazemetostat | ||||||
| TSC1, TSC2, or PI3K/mTOR gene mutation | Samotolisib | ||||||
| activating MAPK pathway gene mutation | Selumetinib | ||||||
| ALK or ROS1 gene alteration | Ensartinib | ||||||
| BRAF V600 gene mutation | Vemurafenib | ||||||
| ATM, BRCA1, BRCA2, RAD51C, RAD51D mutations | Olaparib | ||||||
| Rb positive, alterations in cell cycle genes | Palbociclib | ||||||
| MAPK pathway mutations | Ulixertinib | ||||||
| HRAS gene alterations | Tipifarnib | ||||||
| RET activating mutations | Selpercatinib | ||||||
| TAPISTRY Platform Study | NCT04589845 (A) | II | ROS1 fusion | Entrectinib | Solid Tumor | all | 770 |
| NTRK1/2/3 fusion | Entrectinib | ||||||
| ALK fusion | Alectinib | ||||||
| AKT1/2/3 mutation | Ipatasertib | ||||||
| PIK3CA multiple mutation | Inavolisib | ||||||
| BRAF mutation or fusion-positive | Belvarafenib | ||||||
| RET fusion-positive | Pralsetinib |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suthapot, P.; Chiangjong, W.; Chaiyawat, P.; Choochuen, P.; Pruksakorn, D.; Sangkhathat, S.; Hongeng, S.; Anurathapan, U.; Chutipongtanate, S. Genomics-Driven Precision Medicine in Pediatric Solid Tumors. Cancers 2023, 15, 1418. https://doi.org/10.3390/cancers15051418
Suthapot P, Chiangjong W, Chaiyawat P, Choochuen P, Pruksakorn D, Sangkhathat S, Hongeng S, Anurathapan U, Chutipongtanate S. Genomics-Driven Precision Medicine in Pediatric Solid Tumors. Cancers. 2023; 15(5):1418. https://doi.org/10.3390/cancers15051418
Chicago/Turabian StyleSuthapot, Praewa, Wararat Chiangjong, Parunya Chaiyawat, Pongsakorn Choochuen, Dumnoensun Pruksakorn, Surasak Sangkhathat, Suradej Hongeng, Usanarat Anurathapan, and Somchai Chutipongtanate. 2023. "Genomics-Driven Precision Medicine in Pediatric Solid Tumors" Cancers 15, no. 5: 1418. https://doi.org/10.3390/cancers15051418
APA StyleSuthapot, P., Chiangjong, W., Chaiyawat, P., Choochuen, P., Pruksakorn, D., Sangkhathat, S., Hongeng, S., Anurathapan, U., & Chutipongtanate, S. (2023). Genomics-Driven Precision Medicine in Pediatric Solid Tumors. Cancers, 15(5), 1418. https://doi.org/10.3390/cancers15051418