

Deregulated Gene Expression Profiles and Regulatory Networks in Adult and Pediatric RUNX1/RUNX1T1-Positive AML Patients


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Extraction and Gene Expression Analysis in RUNX1/RUNX1T1 AML Patients
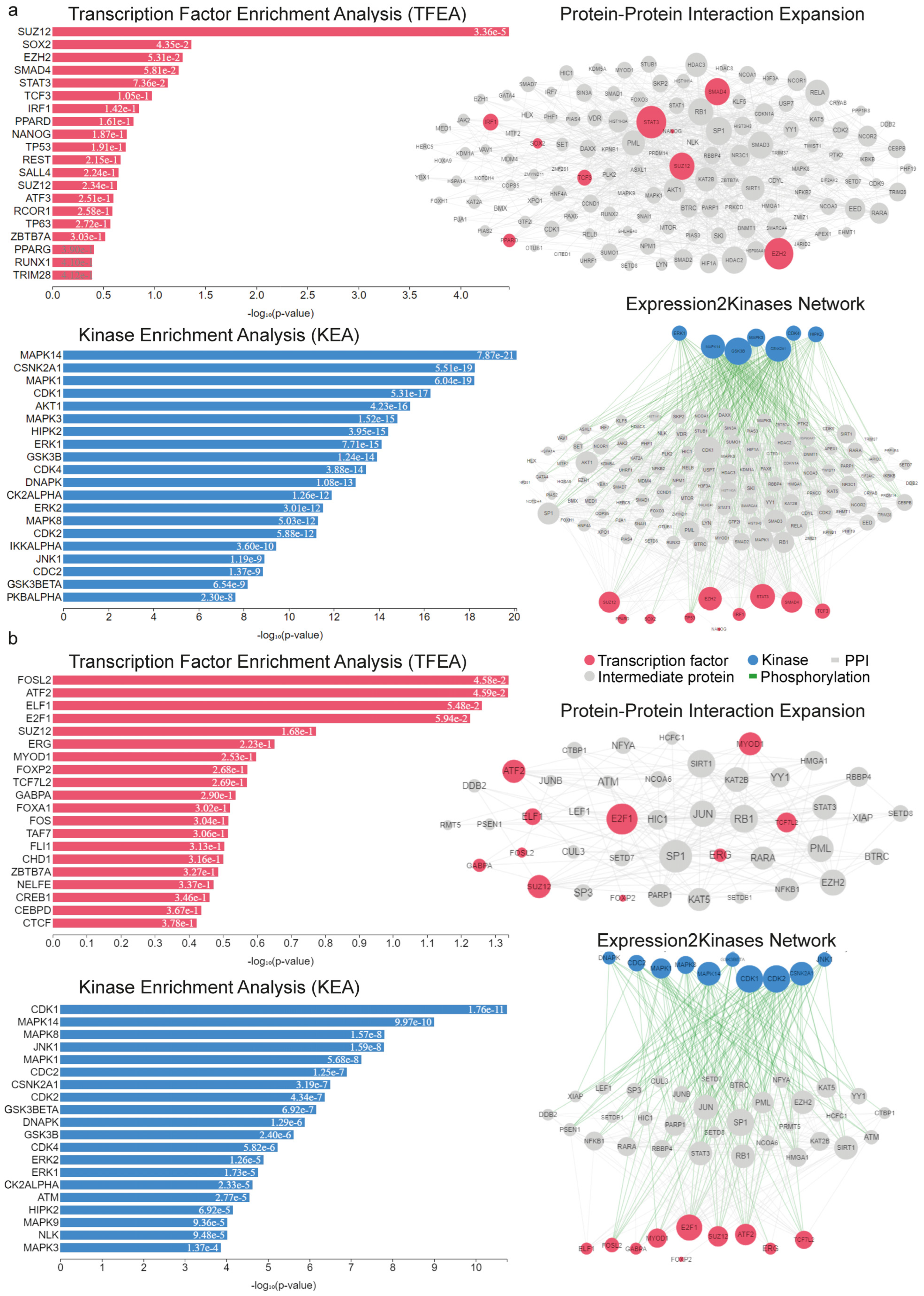
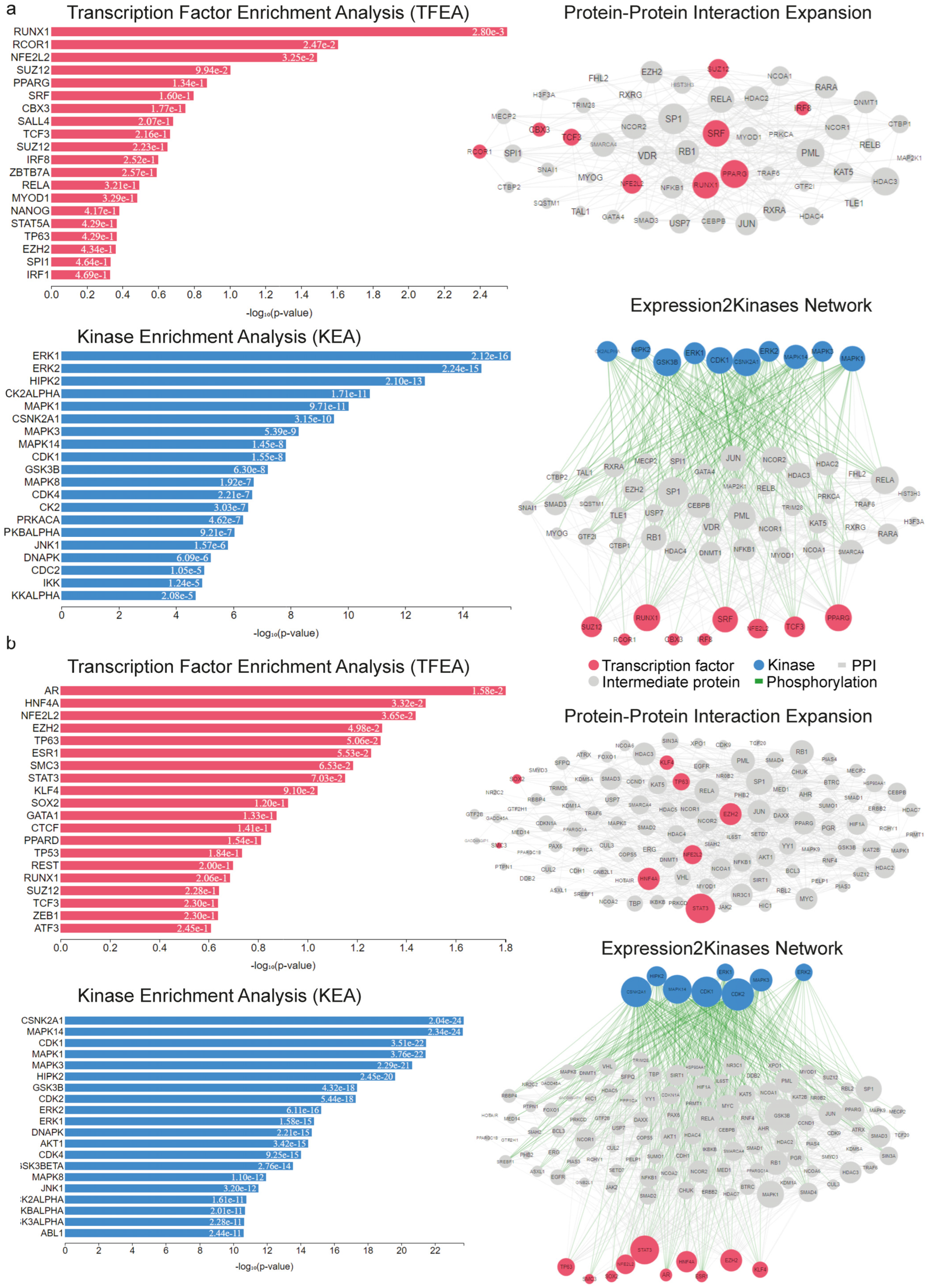
2.2. Upstream Regulators of the Deregulated Genes in RUNX1/RUNX1T1 AML Patients
2.3. Statistical Analysis
3. Results
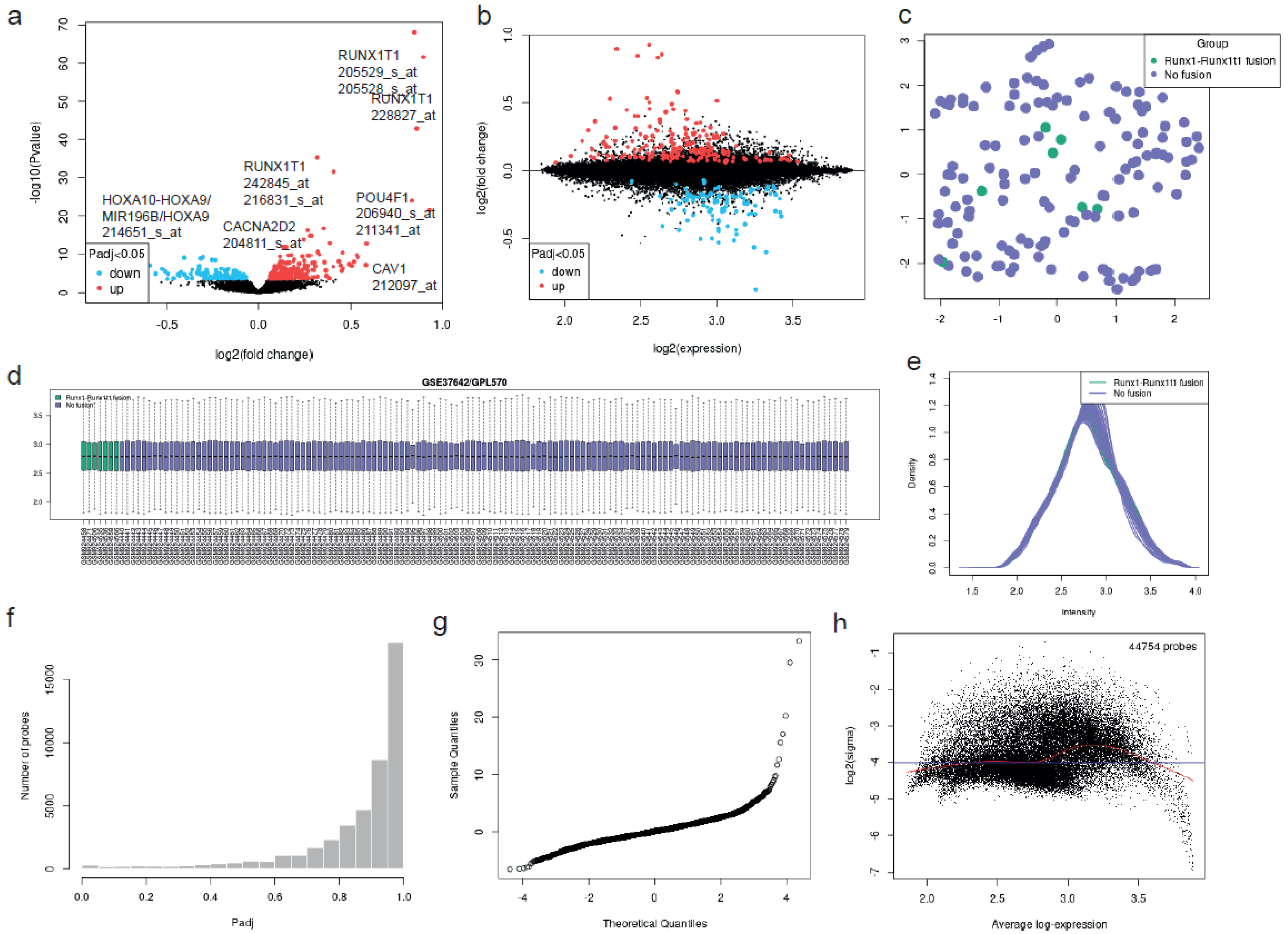
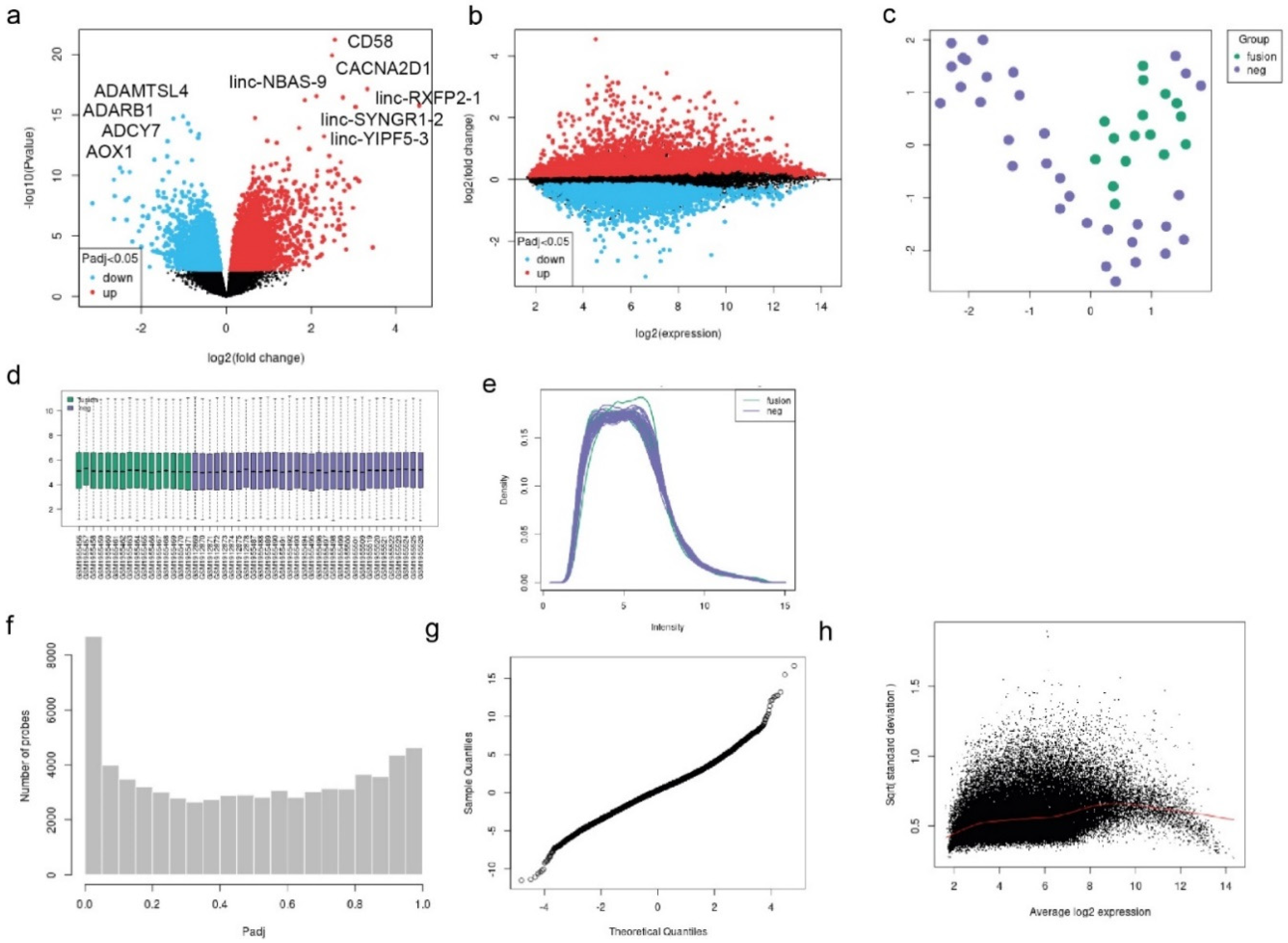
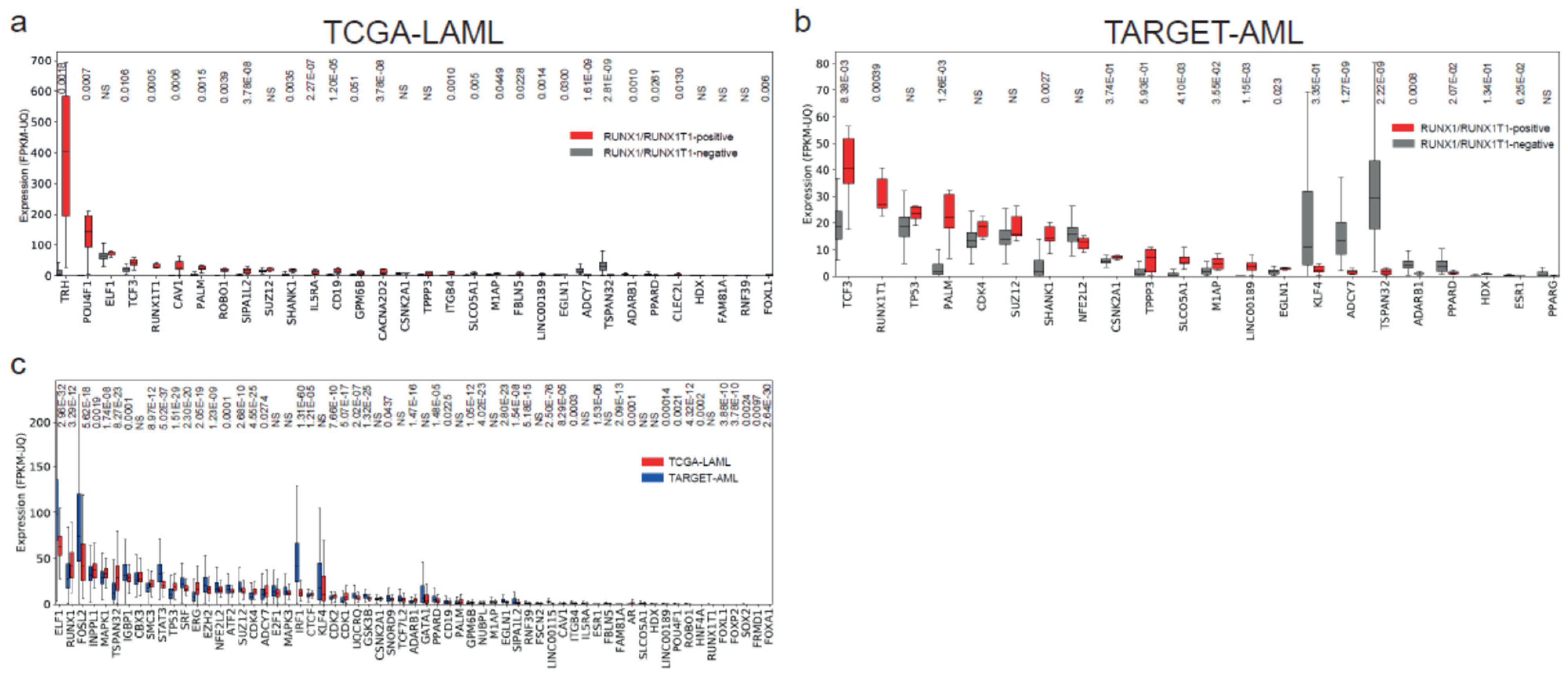
Deregulated Genes and Functional Analysis in RUNX1/RUNX1T1 AML Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, J.; Wang, S.; Hong, S.; Hong, S.; He, S. The Epidemiological Trend of Acute Myeloid Leukemia in Childhood: A Population-Based Analysis. J. Cancer 2019, 10, 4824–4835. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [Green Version]
- Odenike, O.M.; Alkan, S.; Sher, D.; Godwin, J.E.; Huo, D.; Brandt, S.J.; Green, M.; Xie, J.; Zhang, Y.; Vesole, D.H.; et al. Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clin. Cancer Res. 2008, 14, 7095–7101. [Google Scholar] [CrossRef] [Green Version]
- Kunadt, D.; Dransfeld, C.; Dill, C.; Schmiedgen, M.; Kramer, M.; Altmann, H.; Röllig, C.; Bornhäuser, M.; Mahlknecht, U.; Schaich, M.; et al. Multidrug-related protein 1 (MRP1) polymorphisms rs129081, rs212090, and rs212091 predict survival in normal karyotype acute myeloid leukemia. Ann. Hematol. 2020, 99, 2173–2180. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Labbozzetta, M.; Barreca, M.; Spanò, V.; Raimondi, M.V.; Poma, P.; Notarbartolo, M.; Barraja, P.; Montalbano, A. Novel insights on [1,2]oxazolo[5,4-e]isoindoles on multidrug resistant acute myeloid leukemia cell line. Drug Dev. Res. 2022, 83, 1331–1341. [Google Scholar] [CrossRef]
- Fujimoto, T.; Anderson, K.; Jacobsen, S.E.W.; Nishikawa, S.-I.; Nerlov, C. Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPalpha interaction. EMBO J. 2007, 26, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.; Frank, R.C.; Zhang, J.; Miyazaki, Y.; Nimer, S.D. Functional and physical interactions between AML1 proteins and an ETS protein, MEF: Implications for the pathogenesis of t(8;21)-positive leukemias. Mol. Cell. Biol. 1999, 19, 3635–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.-Y.; Akbarali, Y.; Zerbini, L.F.; Gu, X.; Boltax, J.; Wang, Y.; Oettgen, P.; Zhang, D.-E.; Libermann, T.A. Isoforms of the Ets transcription factor NERF/ELF-2 physically interact with AML1 and mediate opposing effects on AML1-mediated transcription of the B cell-specific blk gene. J. Biol. Chem. 2004, 279, 19512–19522. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, D.G.; Jordan, C.T.; Felix, C.A. The molecular basis of leukemia. Hematology 2004, 2004, 80–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucci, G.; Mrózek, K.; Ruppert, A.S.; Maharry, K.; Kolitz, J.E.; Moore, J.O.; Mayer, R.J.; Pettenati, M.J.; Powell, B.L.; Edwards, C.G.; et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): A Cancer and Leukemia Group B study. J. Clin. Oncol. 2005, 23, 5705–5717. [Google Scholar] [CrossRef] [PubMed]
- Hospital, M.-A.; Prebet, T.; Bertoli, S.; Thomas, X.; Tavernier, E.; Braun, T.; Pautas, C.; Perrot, A.; Lioure, B.; Rousselot, P.; et al. Core-binding factor acute myeloid leukemia in first relapse: A retrospective study from the French AML Intergroup. Blood 2014, 124, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Li, W. The 5th Edition of the World Health Organization Classification of Hematolymphoid Tumors. In Leukemia; Li, W., Ed.; Exon Publications: Brisbane, Australia, 2022; ISBN 978-0-645-33207-0. [Google Scholar]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K.; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, K.; Kaspers, G.; Harrison, C.J.; Beverloo, H.B.; Reedijk, A.; Bongers, M.; Cloos, J.; Pession, A.; Reinhardt, D.; Zimmerman, M.; et al. Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J. Clin. Oncol. 2015, 33, 4247–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandoli, A.; Singh, A.A.; Prange, K.H.M.; Tijchon, E.; Oerlemans, M.; Dirks, R.; Ter Huurne, M.; Wierenga, A.T.J.; Janssen-Megens, E.M.; Berentsen, K.; et al. The Hematopoietic Transcription Factors RUNX1 and ERG Prevent AML1-ETO Oncogene Overexpression and Onset of the Apoptosis Program in t(8;21) AMLs. Cell Rep. 2016, 17, 2087–2100. [Google Scholar] [CrossRef] [Green Version]
- Ptasinska, A.; Assi, S.A.; Martinez-Soria, N.; Imperato, M.R.; Piper, J.; Cauchy, P.; Pickin, A.; James, S.R.; Hoogenkamp, M.; Williamson, D.; et al. Identification of a dynamic core transcriptional network in t(8;21) AML that regulates differentiation block and self-renewal. Cell Rep. 2014, 8, 1974–1988. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell 2018, 34, 626–642.e8. [Google Scholar] [CrossRef] [Green Version]
- Grinev, V.V.; Barneh, F.; Ilyushonak, I.M.; Nakjang, S.; Smink, J.; van Oort, A.; Clough, R.; Seyani, M.; McNeill, H.; Reza, M.; et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat. Commun. 2021, 12, 520. [Google Scholar] [CrossRef]
- Herold, T.; Jurinovic, V.; Batcha, A.M.N.; Bamopoulos, S.A.; Rothenberg-Thurley, M.; Ksienzyk, B.; Hartmann, L.; Greif, P.A.; Phillippou-Massier, J.; Krebs, S.; et al. A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica 2018, 103, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Herold, T.; Metzeler, K.H.; Vosberg, S.; Hartmann, L.; Röllig, C.; Stölzel, F.; Schneider, S.; Hubmann, M.; Zellmeier, E.; Ksienzyk, B.; et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood 2014, 124, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Herold, T.; He, C.; Valk, P.J.M.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.D.; Maharry, K.S.; Sun, M.; et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: An international collaborative study. J. Clin. Oncol. 2013, 31, 1172–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuett, A.; Rieger, C.; Perathoner, D.; Herold, T.; Wagner, M.; Sironi, S.; Sotlar, K.; Horny, H.-P.; Deniffel, C.; Drolle, H.; et al. IL-8 as mediator in the microenvironment-leukaemia network in acute myeloid leukaemia. Sci. Rep. 2015, 5, 18411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tregnago, C.; Manara, E.; Zampini, M.; Bisio, V.; Borga, C.; Bresolin, S.; Aveic, S.; Germano, G.; Basso, G.; Pigazzi, M. CREB engages C/EBPδ to initiate leukemogenesis. Leukemia 2016, 30, 1887–1896. [Google Scholar] [CrossRef]
- Porcù, E.; Benetton, M.; Bisio, V.; Da Ros, A.; Tregnago, C.; Borella, G.; Zanon, C.; Bordi, M.; Germano, G.; Manni, S.; et al. The long non-coding RNA CDK6-AS1 overexpression impacts on acute myeloid leukemia differentiation and mitochondrial dynamics. iScience 2021, 24, 103350. [Google Scholar] [CrossRef]
- Gundersen, G.W.; Jones, M.R.; Rouillard, A.D.; Kou, Y.; Monteiro, C.D.; Feldmann, A.S.; Hu, K.S.; Ma’ayan, A. GEO2Enrichr: Browser extension and server app to extract gene sets from GEO and analyze them for biological functions. Bioinformatics 2015, 31, 3060–3062. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G.; et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016, 7, 12846. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altered Transcriptome in Pediatric AML Compared with NormalHematopoiesis. Br. J. Cancer Res. 2020, 3. [CrossRef]
- The Cancer Genome Atlas Research Network Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Brettingham-Moore, K.H.; Taberlay, P.C.; Holloway, A.F. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front. Immunol. 2015, 6, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Harbi, S.; Aljurf, M.; Mohty, M.; Almohareb, F.; Ahmed, S.O.A. An update on the molecular pathogenesis and potential therapeutic targeting of AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1. Blood Adv. 2020, 4, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, K.; Zhang, D.-E. RUNX1 and RUNX1-ETO: Roles in hematopoiesis and leukemogenesis. Front. Biosci. (Landmark Ed) 2012, 17, 1120–1139. [Google Scholar] [CrossRef] [Green Version]
- Melnick, A.M.; Westendorf, J.J.; Polinger, A.; Carlile, G.W.; Arai, S.; Ball, H.J.; Lutterbach, B.; Hiebert, S.W.; Licht, J.D. The ETO protein disrupted in t(8;21)-associated acute myeloid leukemia is a corepressor for the promyelocytic leukemia zinc finger protein. Mol. Cell. Biol. 2000, 20, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Rochford, J.J.; Semple, R.K.; Laudes, M.; Boyle, K.B.; Christodoulides, C.; Mulligan, C.; Lelliott, C.J.; Schinner, S.; Hadaschik, D.; Mahadevan, M.; et al. ETO/MTG8 is an inhibitor of C/EBPbeta activity and a regulator of early adipogenesis. Mol. Cell. Biol. 2004, 24, 9863–9872. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.N.; McGhee, L.; Meyers, S. The ETO (MTG8) gene family. Gene 2003, 303, 1–10. [Google Scholar] [CrossRef]
- Tighe, J.E.; Calabi, F. t(8;21) breakpoints are clustered between alternatively spliced exons of MTG8. Clin. Sci. 1995, 89, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Swart, L.E.; Heidenreich, O. The RUNX1/RUNX1T1 network: Translating insights into therapeutic options. Exp. Hematol. 2021, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; Zhang, J.; Fanelli, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol. Cell. Biol. 1998, 18, 7185–7191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangala, R.K.; Heiss-Neumann, M.S.; Rangatia, J.S.; Singh, S.M.; Schoch, C.; Tenen, D.G.; Hiddemann, W.; Behre, G. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 2003, 101, 270–277. [Google Scholar] [CrossRef]
- Choi, Y.; Elagib, K.E.; Delehanty, L.L.; Goldfarb, A.N. Erythroid inhibition by the leukemic fusion AML1-ETO is associated with impaired acetylation of the major erythroid transcription factor GATA-1. Cancer Res. 2006, 66, 2990–2996. [Google Scholar] [CrossRef] [Green Version]
- Pabst, T.; Mueller, B.U.; Harakawa, N.; Schoch, C.; Haferlach, T.; Behre, G.; Hiddemann, W.; Zhang, D.E.; Tenen, D.G. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat. Med. 2001, 7, 444–451. [Google Scholar] [CrossRef]
- Cheng, C.K.; Li, L.; Cheng, S.H.; Lau, K.M.; Chan, N.P.H.; Wong, R.S.M.; Shing, M.M.K.; Li, C.K.; Ng, M.H.L. Transcriptional repression of the RUNX3/AML2 gene by the t(8;21) and inv(16) fusion proteins in acute myeloid leukemia. Blood 2008, 112, 3391–3402. [Google Scholar] [CrossRef]
- Yang, G.; Khalaf, W.; van de Locht, L.; Jansen, J.H.; Gao, M.; Thompson, M.A.; van der Reijden, B.A.; Gutmann, D.H.; Delwel, R.; Clapp, D.W.; et al. Transcriptional repression of the Neurofibromatosis-1 tumor suppressor by the t(8;21) fusion protein. Mol. Cell. Biol. 2005, 25, 5869–5879. [Google Scholar] [CrossRef] [Green Version]
- Vegi, N.M.; Klappacher, J.; Oswald, F.; Mulaw, M.A.; Mandoli, A.; Thiel, V.N.; Bamezai, S.; Feder, K.; Martens, J.H.A.; Rawat, V.P.S.; et al. MEIS2 Is an Oncogenic Partner in AML1-ETO-Positive AML. Cell Rep. 2016, 16, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Klampfer, L.; Zhang, J.; Zelenetz, A.O.; Uchida, H.; Nimer, S.D. The AML1/ETO fusion protein activates transcription of BCL-2. Proc. Natl. Acad. Sci. USA 1996, 93, 14059–14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, F.-S.; Griesinger, A.; Wunderlich, M.; Lin, S.; Link, K.A.; Shrestha, M.; Goyama, S.; Mizukawa, B.; Shen, S.; Marcucci, G.; et al. The thrombopoietin/MPL/Bcl-xL pathway is essential for survival and self-renewal in human preleukemia induced by AML1-ETO. Blood 2012, 120, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.-S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Meier, P.J. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta 2003, 1609, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Asp. Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, N.; Lockhart, A.C.; Lee, W. Role of Organic Anion-Transporting Polypeptides (OATPs) in Cancer Therapy. AAPS J. 2015, 17, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Fortier, J.M.; Payton, J.E.; Cahan, P.; Ley, T.J.; Walter, M.J.; Graubert, T.A. POU4F1 is associated with t(8;21) acute myeloid leukemia and contributes directly to its unique transcriptional signature. Leukemia 2010, 24, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef]
- Navarro, A.; Anand-Apte, B.; Parat, M.-O. A role for caveolae in cell migration. FASEB J. 2004, 18, 1801–1811. [Google Scholar] [CrossRef]
- Caliceti, C.; Zambonin, L.; Rizzo, B.; Fiorentini, D.; Vieceli Dalla Sega, F.; Hrelia, S.; Prata, C. Role of plasma membrane caveolae/lipid rafts in VEGF-induced redox signaling in human leukemia cells. BioMed. Res. Int. 2014, 2014, 857504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xie, J.; Lu, Z.; Chen, C.; Yin, Y.; Zhan, R.; Fang, Y.; Hu, X.; Zhang, C.C. ADCY7 supports development of acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2015, 465, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.-H.; Chong, J.-H.; Guo, Y.; Zeng, H.-M.; Liu, S.-Y.; Xu, L.-L.; Wei, J.; Lin, Y.-M.; Zhu, X.-F.; Zheng, G.-G. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem. Biophys. Res. Commun. 2011, 406, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xu, Z.; Huang, M.; Wang, X.; Ren, X.; Cai, Y.; Peng, B.; Liang, Q.; Chen, X.; Yan, Y. Downregulated ADARB1 Facilitates Cell Proliferation, Invasion and has Effect on the Immune Regulation in Ovarian Cancer. Front. Bioeng. Biotechnol. 2021, 9, 792911. [Google Scholar] [CrossRef] [PubMed]
- XuFeng, R.; Boyer, M.J.; Shen, H.; Li, Y.; Yu, H.; Gao, Y.; Yang, Q.; Wang, Q.; Cheng, T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc. Natl. Acad. Sci. USA 2009, 106, 17763–17768. [Google Scholar] [CrossRef] [Green Version]
- Maiga, A.; Lemieux, S.; Pabst, C.; Lavallée, V.-P.; Bouvier, M.; Sauvageau, G.; Hébert, J. Transcriptome analysis of G protein-coupled receptors in distinct genetic subgroups of acute myeloid leukemia: Identification of potential disease-specific targets. Blood Cancer J. 2016, 6, e431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, W.M.; Currid, C.A.; Whelan, L.C. Fibulins and cancer: Friend or foe? Trends Mol. Med. 2005, 11, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, X.; Yue, W.; Chen, D.; Yu, J.; Yao, Z.; Zhang, L. Fibulin-5 inhibits Wnt/β-catenin signaling in lung cancer. Oncotarget 2015, 6, 15022–15034. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Sun, Q.; Landreneau, R.; Wu, C.; Siegfried, J.M.; Yu, J.; Zhang, L. Fibulin-5 suppresses lung cancer invasion by inhibiting matrix metalloproteinase-7 expression. Cancer Res. 2009, 69, 6339–6346. [Google Scholar] [CrossRef] [Green Version]
- Mohamedi, Y.; Fontanil, T.; Solares, L.; Garcia-Suárez, O.; García-Piqueras, J.; Vega, J.A.; Cal, S.; Obaya, A.J. Fibulin-5 downregulates Ki-67 and inhibits proliferation and invasion of breast cancer cells. Int. J. Oncol. 2016, 48, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Tu, K.; Dou, C.; Zheng, X.; Li, C.; Yang, W.; Yao, Y.; Liu, Q. Fibulin-5 inhibits hepatocellular carcinoma cell migration and invasion by down-regulating matrix metalloproteinase-7 expression. BMC Cancer 2014, 14, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Kong, F.; Huang, K.; Li, L.; Li, Z.; Wang, X.; Zhang, W.; Wu, X. LncRNA MIR210HG promotes proliferation and invasion of non-small cell lung cancer by upregulating methylation of CACNA2D2 promoter via binding to DNMT1. OncoTargets Ther. 2019, 12, 3779–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zou, Y.; Li, T.; Wong, T.K.F.; Bushey, R.T.; Campa, M.J.; Gottlin, E.B.; Liu, H.; Wei, Q.; Rodrigo, A.; et al. Genetic Variants of CLPP and M1AP Are Associated With Risk of Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 709829. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, X.; Li, J.; Su, Q.; Qiu, Y.; Zhang, Z.; Zhang, L.; Mo, W. TPPP3 Associated with Prognosis and Immune Infiltrates in Head and Neck Squamous Carcinoma. BioMed Res. Int. 2020, 2020, 3962146. [Google Scholar] [CrossRef]
- Hemler, M.E. Targeting of tetraspanin proteins—potential benefits and strategies. Nat. Rev. Drug Discov. 2008, 7, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Sun, H.; Lin, R.; Zhang, J.; Yin, H.; Xian, S.; Li, M.; Wang, S.; Li, Z.; Qiao, Y.; et al. The role of tetraspanins pan-cancer. iScience 2022, 25, 104777. [Google Scholar] [CrossRef]
- Assi, S.A.; Imperato, M.R.; Coleman, D.J.L.; Pickin, A.; Potluri, S.; Ptasinska, A.; Chin, P.S.; Blair, H.; Cauchy, P.; James, S.R.; et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 2019, 51, 151–162. [Google Scholar] [CrossRef]
- Lee, S.C.W.; Miller, S.; Hyland, C.; Kauppi, M.; Lebois, M.; Di Rago, L.; Metcalf, D.; Kinkel, S.A.; Josefsson, E.C.; Blewitt, M.E.; et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 2015, 126, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Sauvageau, M.; Sauvageau, G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Radulović, V.; de Haan, G.; Klauke, K. Polycomb-group proteins in hematopoietic stem cell regulation and hematopoietic neoplasms. Leukemia 2013, 27, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, J.; Xu, L.; Găman, M.-A.; Zou, Z. The genesis and evolution of acute myeloid leukemia stem cells in the microenvironment: From biology to therapeutic targeting. Cell Death Discov. 2022, 8, 397. [Google Scholar] [CrossRef]
- Park, D.J.; Kwon, A.; Cho, B.-S.; Kim, H.-J.; Hwang, K.-A.; Kim, M.; Kim, Y. Characteristics of DNMT3A mutations in acute myeloid leukemia. Blood Res. 2020, 55, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Chu, M.-Q.; Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Ma, J.-C.; Zhang, W.; Wen, X.-M.; Lin, J.; Qian, J.; Zhou, J.-D. EZH2 dysregulation: Potential biomarkers predicting prognosis and guiding treatment choice in acute myeloid leukaemia. J. Cell. Mol. Med. 2020, 24, 1640–1649. [Google Scholar] [CrossRef] [Green Version]
- Visser, H.P.; Gunster, M.J.; Kluin-Nelemans, H.C.; Manders, E.M.; Raaphorst, F.M.; Meijer, C.J.; Willemze, R.; Otte, A.P. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br. J. Haematol. 2001, 112, 950–958. [Google Scholar] [CrossRef]
- Fiskus, W.; Pranpat, M.; Balasis, M.; Herger, B.; Rao, R.; Chinnaiyan, A.; Atadja, P.; Bhalla, K. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol. Cancer Ther. 2006, 5, 3096–3104. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, L.; Li, X.; Chen, F.; Jiang, L.; Yu, G.; Wang, Z.; Yin, C.; Jiang, X.; Zhong, Q.; et al. Higher EZH2 expression is associated with extramedullary infiltration in acute myeloid leukemia. Tumor Biol. 2016, 37, 11409–11420. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.-W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, Ø.; Nepstad, I.; Hauge, M.; Hatfield, K.J.; Reikvam, H. STAT3 as a possible therapeutic target in human malignancies: Lessons from acute myeloid leukemia. Expert Rev. Hematol. 2015, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 signaling in acute myeloid leukemia: Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Aigner, P.; Mizutani, T.; Horvath, J.; Eder, T.; Heber, S.; Lind, K.; Just, V.; Moll, H.P.; Yeroslaviz, A.; Fischer, M.J.M.; et al. STAT3β is a tumor suppressor in acute myeloid leukemia. Blood Adv. 2019, 3, 1989–2002. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 2015, 5, 17663. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-F.; Lai, R. STAT3 in Cancer-Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanellou, P.; Georgakopoulos-Soares, I.; Zaravinos, A. Deregulated Gene Expression Profiles and Regulatory Networks in Adult and Pediatric RUNX1/RUNX1T1-Positive AML Patients. Cancers 2023, 15, 1795. https://doi.org/10.3390/cancers15061795
Kanellou P, Georgakopoulos-Soares I, Zaravinos A. Deregulated Gene Expression Profiles and Regulatory Networks in Adult and Pediatric RUNX1/RUNX1T1-Positive AML Patients. Cancers. 2023; 15(6):1795. https://doi.org/10.3390/cancers15061795
Chicago/Turabian StyleKanellou, Peggy, Ilias Georgakopoulos-Soares, and Apostolos Zaravinos. 2023. "Deregulated Gene Expression Profiles and Regulatory Networks in Adult and Pediatric RUNX1/RUNX1T1-Positive AML Patients" Cancers 15, no. 6: 1795. https://doi.org/10.3390/cancers15061795
APA StyleKanellou, P., Georgakopoulos-Soares, I., & Zaravinos, A. (2023). Deregulated Gene Expression Profiles and Regulatory Networks in Adult and Pediatric RUNX1/RUNX1T1-Positive AML Patients. Cancers, 15(6), 1795. https://doi.org/10.3390/cancers15061795