
NKG2D Fine-Tunes the Local Inflammatory Response in Colorectal Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Lymphocyte Isolation from Intestinal and Tumor Tissue
2.3. Flow Cytometry
2.4. Analysis of TCGA Gene Expression Database
2.4.1. Data Acquisition and Processing
2.4.2. Gene Set Enrichment Analysis (GSEA)
2.5. Statistical Analysis
3. Results
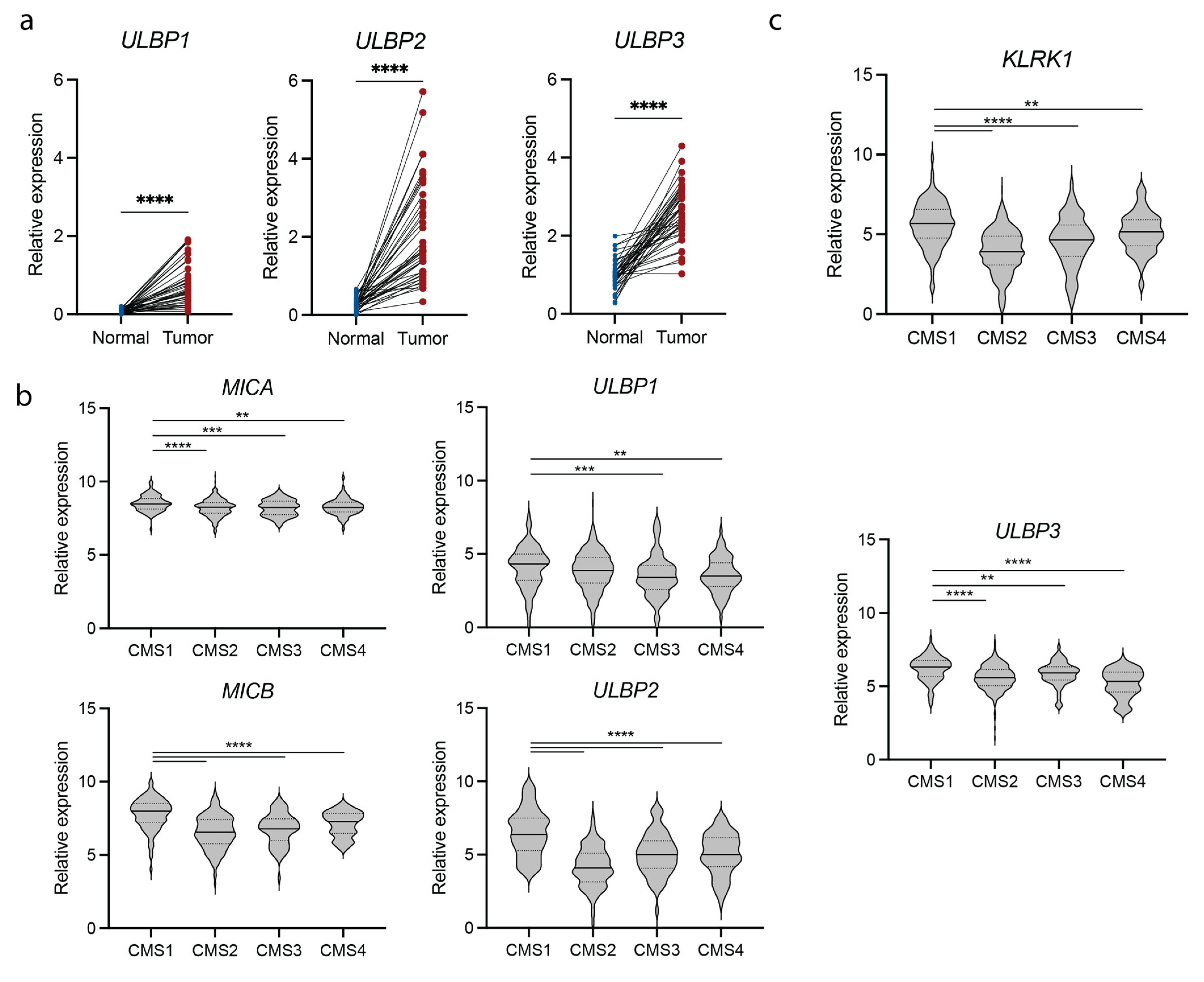
3.1. NKG2D and NKG2D Ligand Expression Is Upregulated in Colorectal Cancer Patients
3.2. NKG2D Expression Associates with an IFNγ Signature
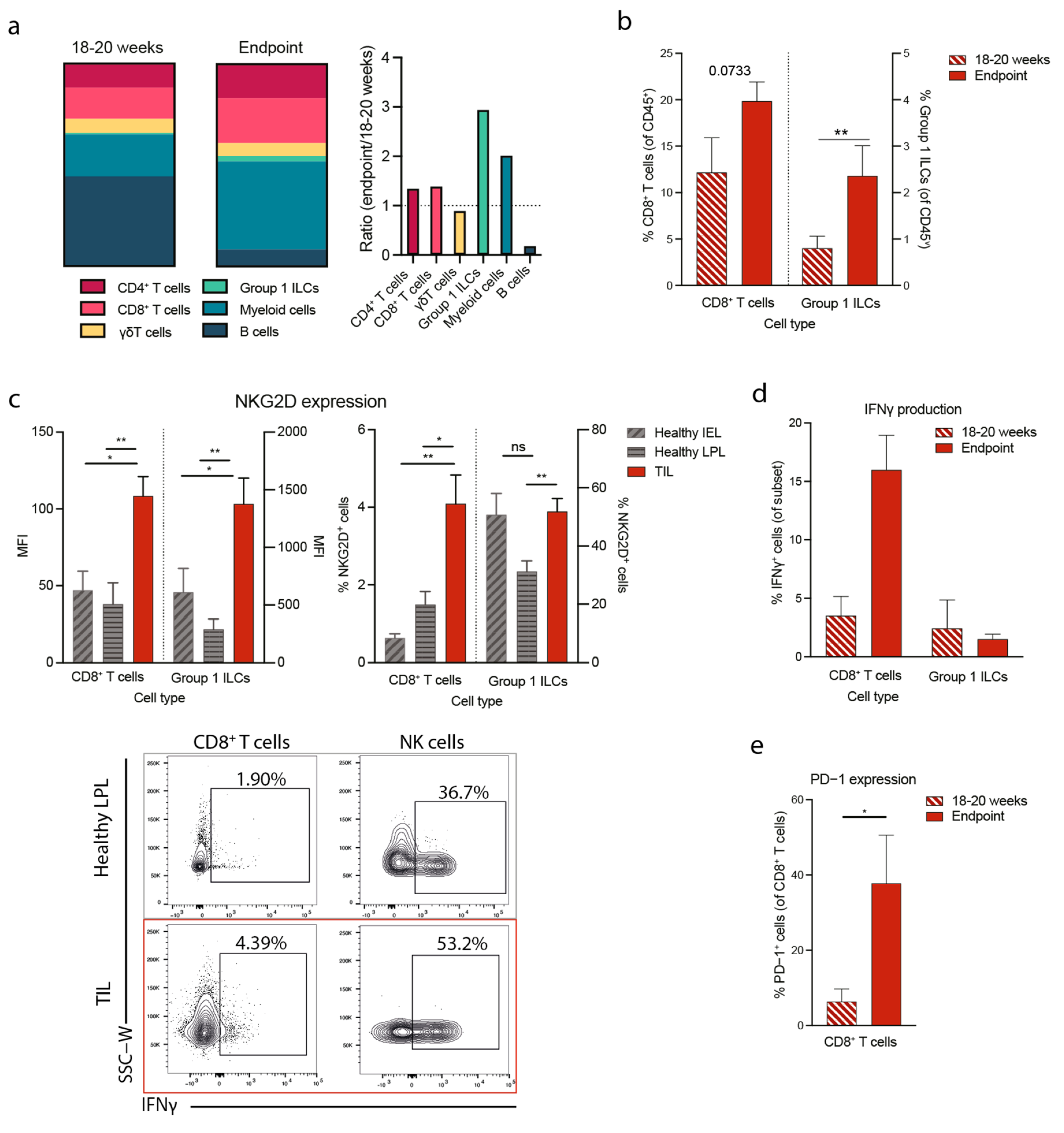
3.3. Tumorigenesis in the Apcmin/+ Mouse Correlates with Increased NKG2D Expression and Type 1 Immunity
3.4. NKG2D Deficiency Reduces IFNγ Production in the Tumor Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Part I: National Cancer Statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Golshani, G.; Zhang, Y. Advances in Immunotherapy for Colorectal Cancer: A Review. Ther. Adv. Gastroenter. 2020, 13, 1756284820917527. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, Tumor Trafficking and Immunogenicity of Chimeric Antigen Receptor (CAR)-T Cells Specific for TAG-72 in Colorectal Cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, X.; Hu, W. Advances in Adoptive Cellular Therapy for Colorectal Cancer: A Narrative Review. Ann. Transl. Med. 2022, 10, 1404. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Li, H.; Yang, C.; Cheng, H.; Huang, S.; Zheng, Y. CAR-T Cells for Colorectal Cancer: Target-Selection and Strategies for Improved Activity and Safety. J. Cancer 2021, 12, 1804–1814. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 575. [Google Scholar] [CrossRef]
- Toledo-Stuardo, K.; Ribeiro, C.H.; Canals, A.; Morales, M.; Gárate, V.; Rodríguez-Siza, J.; Tello, S.; Bustamante, M.; Armisen, R.; Matthies, D.J.; et al. Major Histocompatibility Complex Class I-Related Chain A (MICA) Allelic Variants Associate with Susceptibility and Prognosis of Gastric Cancer. Front. Immunol. 2021, 12, 645528. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Meehan, K.R.; Sentman, C.L. Treatment of Multiple Myeloma with Adoptively Transferred Chimeric NKG2D Receptor-Expressing T Cells. Gene Ther. 2011, 18, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Metais, J.-Y.; Escudero, A.; Vela, M.; Valentín, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patiño-García, A.; et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef]
- Deng, X.; Gao, F.; Li, N.; Li, Q.; Zhou, Y.; Yang, T.; Cai, Z.; Du, P.; Chen, F.; Cai, J. Antitumor Activity of NKG2D CAR-T Cells against Human Colorectal Cancer Cells in Vitro and in Vivo. Am. J. Cancer Res. 2019, 9, 945–958. [Google Scholar] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK Cells: A Promising Cellular Immunotherapy for Cancer. Ebiomedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Frazao, A.; Rethacker, L.; Messaoudene, M.; Avril, M.-F.; Toubert, A.; Dulphy, N.; Caignard, A. NKG2D/NKG2-Ligand Pathway Offers New Opportunities in Cancer Treatment. Front. Immunol. 2019, 10, 661. [Google Scholar] [CrossRef] [Green Version]
- Curio, S.; Jonsson, G.; Marinovic, S. A Summary of Current NKG2D-Based CAR Clinical Trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef]
- Bachier, C.; Borthakur, G.; Hosing, C.; Blum, W.; Rotta, M.; Ojeras, P.; Barnett, B.; Rajangam, K.; Majhail, N.S.; Nikiforow, S. A Phase 1 Study of NKX101, an Allogeneic CAR Natural Killer (NK) Cell Therapy, in Subjects with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) or Higher-Risk Myelodysplastic Syndrome (MDS). Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Michaux, A.; Mauën, S.; Breman, E.; Dheur, M.-S.; Twyffels, L.; Saerens, L.; Jacques-Hespel, C.; Gauthy, E.; Agaugué, S.; Gilham, D.E.; et al. Clinical Grade Manufacture of CYAD-101, a NKG2D-Based, First in Class, Non–Gene-Edited Allogeneic CAR T-Cell Therapy. J. Immunother. 2022, 45, 150–161. [Google Scholar] [CrossRef]
- Sallman, D.A.; Kerre, T.; Havelange, V.; Poiré, X.; Lewalle, P.; Wang, E.S.; Brayer, J.B.; Davila, M.L.; Moors, I.; Machiels, J.-P.; et al. CYAD-01, an Autologous NKG2D-Based CAR T-Cell Therapy, in Relapsed or Refractory Acute Myeloid Leukaemia and Myelodysplastic Syndromes or Multiple Myeloma (THINK): Haematological Cohorts of the Dose Escalation Segment of a Phase 1 Trial. Lancet Haematol. 2022, 10, e191–e202. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Dekervel, J.; Hendlisz, A.; Anguille, S.; Awada, A.; Cerf, E.; Lonez, C.; Breman, E.; Dheur, M.-S.; Alcantar-Orozco, E.; et al. Updated Data from AlloSHRINK Phase I First-in-Human Study Evaluating CYAD-101, an Innovative Non-Gene Edited Allogeneic CAR-T in MCRC. J. Clin. Oncol. 2021, 39, 74. [Google Scholar] [CrossRef]
- VanSeggelen, H.; Hammill, J.A.; Dvorkin-Gheva, A.; Tantalo, D.G.; Kwiecien, J.M.; Denisova, G.F.; Rabinovich, B.; Wan, Y.; Bramson, J.L. T Cells Engineered with Chimeric Antigen Receptors Targeting NKG2D Ligands Display Lethal Toxicity in Mice. Mol. Ther. 2015, 23, 1600–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentman, M.-L.; Murad, J.M.; Cook, W.J.; Wu, M.-R.; Reder, J.; Baumeister, S.H.; Dranoff, G.; Fanger, M.W.; Sentman, C.L. Mechanisms of Acute Toxicity in NKG2D Chimeric Antigen Receptor T Cell–Treated Mice. J. Immunol. 2016, 197, 4674–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Powell, D.J. Strain-Dependent Lethal Toxicity in NKG2D Ligand-Targeted CAR T-Cell Therapy. Mol. Ther. 2015, 23, 1559–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.; Douay, C.; et al. CD4+NKG2D+ T Cells in Crohn’s Disease Mediate Inflammatory and Cytotoxic Responses Through MICA Interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Agarwal, S.; Sakiyama, M.; Shenoy, V.; Orr, W.S.; Diffalha, S.A.; Prizment, A.; Varambally, S.; Manne, U.; Gomez, C.R. Expression of MHC Class I Polypeptide-Related Sequence A (MICA) in Colorectal Cancer. Front. Biosci-landmrk 2021, 26, 765. [Google Scholar] [CrossRef]
- Sheppard, S.; Guedes, J.; Mroz, A.; Zavitsanou, A.-M.; Kudo, H.; Rothery, S.M.; Angelopoulos, P.; Goldin, R.; Guerra, N. The Immunoreceptor NKG2D Promotes Tumour Growth in a Model of Hepatocellular Carcinoma. Nat. Commun. 2017, 8, 13930. [Google Scholar] [CrossRef]
- Sheppard, S.; Ferry, A.; Guedes, J.; Guerra, N. The Paradoxical Role of NKG2D in Cancer Immunity. Front. Immunol. 2018, 9, 1808. [Google Scholar] [CrossRef]
- Cadoux, M.; Caruso, S.; Pham, S.; Gougelet, A.; Pophillat, C.; Rioud, R.; Loesch, R.; Colnot, S.; Nguyen, C.T.; Calderaro, J.; et al. Expression of NKG2D Ligands Is Downregulated by Beta-Catenin Signaling and Associates with HCC Aggressiveness. J. Hepatol. 2021, 74, 1386–1397. [Google Scholar] [CrossRef]
- Curio, S.; Edwards, S.C.; Suzuki, T.; McGovern, J.; Triulzi, C.; Yoshida, N.; Jonsson, G.; Glauner, T.; Rami, D.; Wiesheu, R.; et al. NKG2D Signaling Regulates IL-17A-Producing ΓδT Cells in Mice to Promote Cancer Progression. Discov. Immunol. 2022, 1, kyac002. [Google Scholar] [CrossRef]
- Moser, A.R.; Luongo, C.; Gould, K.A.; McNeley, M.K.; Shoemaker, A.R.; Dove, W.F. ApcMin: A Mouse Model for Intestinal and Mammary Tumorigenesis. Eur. J. Cancer 1995, 31, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Mori, H. Multistep Carcinogenesis of the Colon in ApcMin/+ Mouse. Cancer Sci. 2007, 98, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.-T.; Xie, H.-L.; Zhu, L.-C.; Ge, Y.-Z.; Yan, L.; Liao, C.; Gong, Y.-Z.; Shi, H.-P. Immune ULBP1 Is Elevated in Colon Adenocarcinoma and Predicts Prognosis. Front. Genet. 2022, 13, 762514. [Google Scholar] [CrossRef]
- McGilvray, R.W.; Eagle, R.A.; Watson, N.F.S.; Al-Attar, A.; Ball, G.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. NKG2D Ligand Expres-sion in Human Colorectal Cancer Reveals Associations with Prognosis and Evidence for Immunoediting. Clin. Cancer Res. 2009, 15, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, N.F.S.; Spendlove, I.; Madjd, Z.; McGilvray, R.; Green, A.R.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Expression of the Stress-Related MHC Class I Chain-Related Protein MICA Is an Indicator of Good Prognosis in Colorectal Cancer Patients. Int. J. Cancer 2006, 118, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Roelands, J.; Andersen, M.N.; Wieringa, C.H.L.A.; Tollenaar, R.A.E.M.; Hendrickx, W.; Bedognetti, D.; Hokland, M.; Kuppen, P.J.K. Expression of NK Cell Receptor Ligands in Primary Colorectal Cancer Tissue in Relation to the Phenotype of Circulating NK- and NKT Cells, and Clinical Outcome. Mol. Immunol. 2020, 128, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Miranda, L.A.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of Human Colorectal Tumor Spheroids with Immune Cells Reveal the Therapeutic Potential of MICA/B and NKG2A Targeting for Cancer Treatment. J. ImmunoTherapy Cancer 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Victor, C.T.-S.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Xu, B.; Ye, D.; Ren, D.; Wang, S.; Benci, J.L.; Xu, Y.; Ishwaran, H.; Beltra, J.-C.; Wherry, E.J.; et al. Cancer Cells Resistant to Immune Checkpoint Blockade Acquire Interferon-Associated Epigenetic Memory to Sustain T Cell Dysfunction. Nat. Cancer 2023, 4, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Hix, L.M.; Karavitis, J.; Khan, M.W.; Shi, Y.H.; Khazaie, K.; Zhang, M. Tumor STAT1 Transcription Factor Activity Enhances Breast Tumor Growth and Immune Suppression Mediated by Myeloid-Derived Suppressor Cells. J. Biological. Chem. 2013, 288, 11676–11688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Khatun, A.; Kasmani, M.Y.; Chen, Y.; Zheng, S.; Atkinson, S.; Nguyen, C.; Burns, R.; Taparowsky, E.J.; Salzman, N.H.; et al. Group 3 Innate Lymphoid Cells Require BATF to Regulate Gut Homeostasis in Mice. J. Exp. Med. 2022, 219, e20211861. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of Inflammation Is a Risk Factor for Colorectal Neoplasia in Ulcerative Colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curio, S.; Lin, W.; Bromley, C.; McGovern, J.; Triulzi, C.; Jonsson, G.; Ghislat, G.; Zelenay, S.; Guerra, N. NKG2D Fine-Tunes the Local Inflammatory Response in Colorectal Cancer. Cancers 2023, 15, 1792. https://doi.org/10.3390/cancers15061792
Curio S, Lin W, Bromley C, McGovern J, Triulzi C, Jonsson G, Ghislat G, Zelenay S, Guerra N. NKG2D Fine-Tunes the Local Inflammatory Response in Colorectal Cancer. Cancers. 2023; 15(6):1792. https://doi.org/10.3390/cancers15061792
Chicago/Turabian StyleCurio, Sophie, Wanzun Lin, Christian Bromley, Jenny McGovern, Chiara Triulzi, Gustav Jonsson, Ghita Ghislat, Santiago Zelenay, and Nadia Guerra. 2023. "NKG2D Fine-Tunes the Local Inflammatory Response in Colorectal Cancer" Cancers 15, no. 6: 1792. https://doi.org/10.3390/cancers15061792
APA StyleCurio, S., Lin, W., Bromley, C., McGovern, J., Triulzi, C., Jonsson, G., Ghislat, G., Zelenay, S., & Guerra, N. (2023). NKG2D Fine-Tunes the Local Inflammatory Response in Colorectal Cancer. Cancers, 15(6), 1792. https://doi.org/10.3390/cancers15061792