
Precision Oncology: Evolving Clinical Trials across Tumor Types

 , ,
, ,
Abstract
:Simple Summary
Abstract
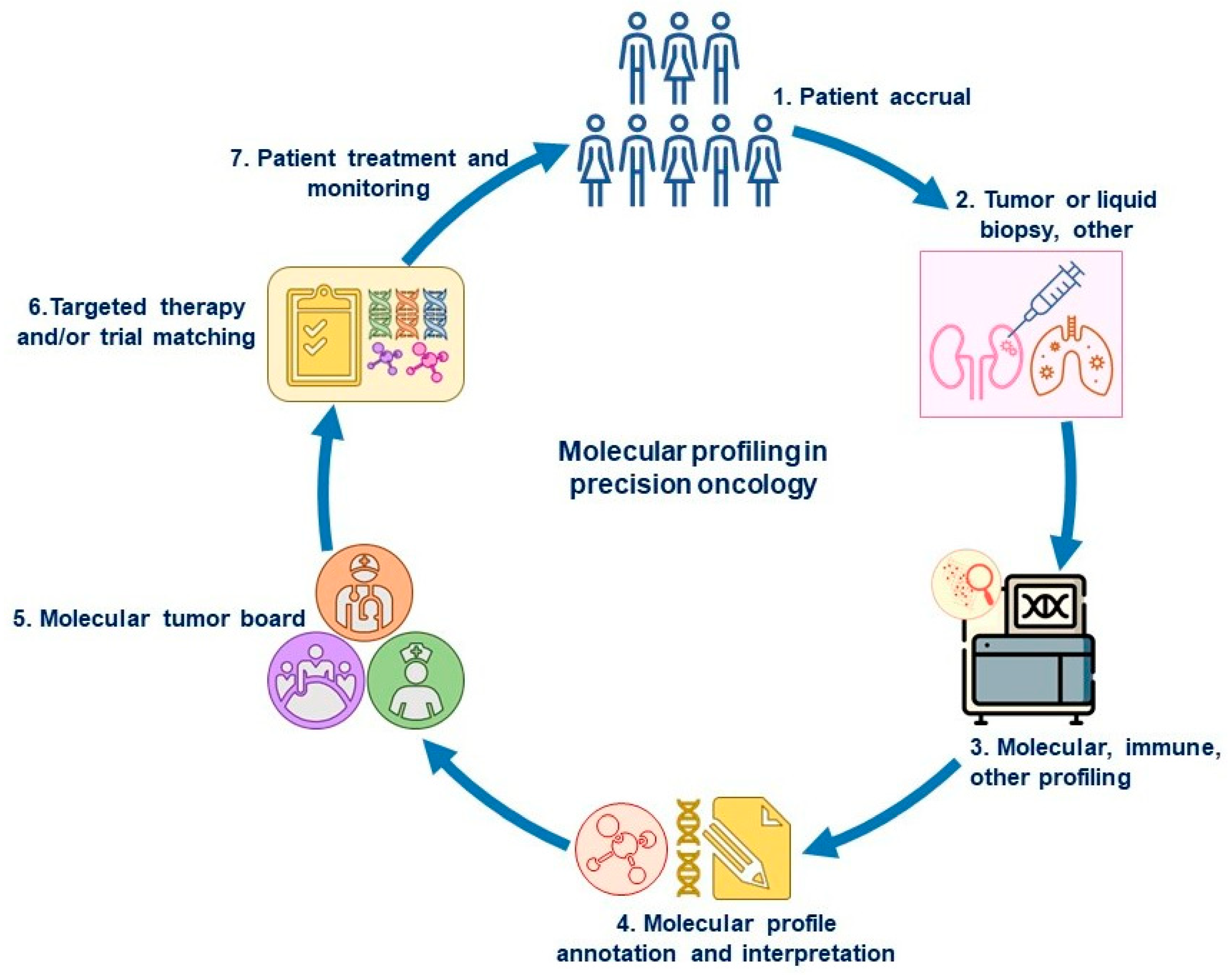
1. Introduction
2. Biomarker-Selected Clinical Trials
2.1. Biomarker Nomenclature, Hierarchy, and Reporting Format
2.2. Biomarker and Literature Evolution
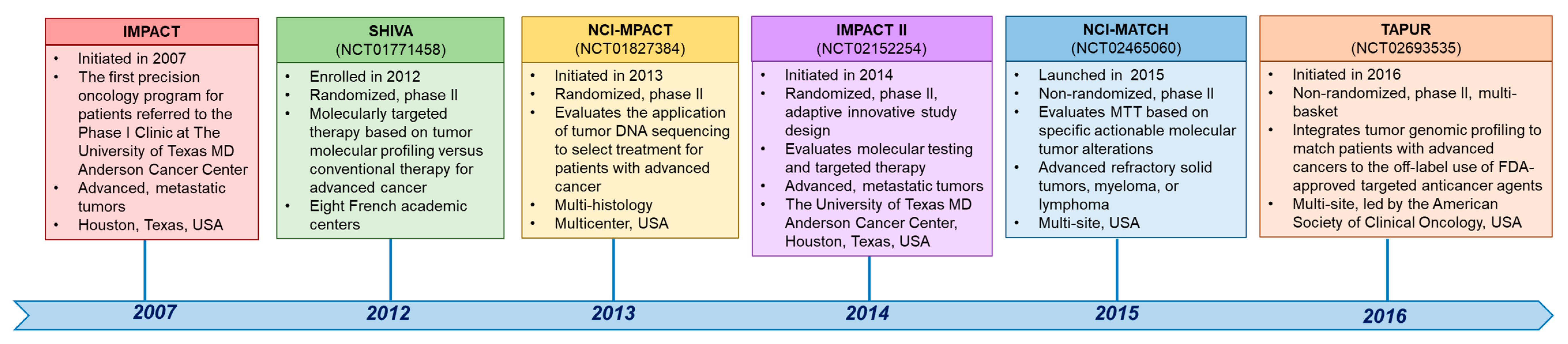
3. Clinical Trials in Precision Oncology across Tumor Types
3.1. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT)
3.2. SHIVA, a Study of Randomized, Molecularly Targeted Therapy Based on Tumor Molecular Profiling versus Conventional Therapy for Advanced Cancer
3.3. Initiative for Molecular Profiling and Advanced Cancer Therapy II (IMPACT2)
3.4. National Cancer Institute Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT)
3.5. The National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI-MATCH)
3.6. Targeted Agent and Profiling Utilization Registry (TAPUR)
3.7. The Drug Rediscovery Protocol (DRUP) Trial
3.8. Other Clinical Trials Focusing on Advanced Diverse Cancers
4. Future Trials
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Johnson, A.; Shufean, M.A.; Kahle, M.; Yang, D.; Woodman, S.E.; Vu, T.; Moorthy, S.; Holla, V.; Meric-Bernstam, F. Operationalization of Next-Generation Sequencing and Decision Support for Precision Oncology. JCO Clin. Cancer Inf. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Fountzilas, E.; Tsimberidou, A.M.; Vo, H.H.; Kurzrock, R. Clinical trial design in the era of precision medicine. Genome Med. 2022, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Stephenson, J.J., Jr.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: Validation and landmark analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M.; Hong, D.S.; Ye, Y.; Cartwright, C.; Wheler, J.J.; Falchook, G.S.; Naing, A.; Fu, S.; Piha-Paul, S.; Janku, F.; et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. 2017, 1, 1–18. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Janku, F.; Naing, A.; Fu, S.; Piha-Paul, S.; Cartwright, C.; Broaddus, R.R.; et al. Long-term overall survival and prognostic score predicting survival: The IMPACT study in precision medicine. J. Hematol. Oncol. 2019, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Brana, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Patel, S.P.; Othus, M.; Chae, Y.K.; Giles, F.J.; Hansel, D.E.; Singh, P.P.; Fontaine, A.; Shah, M.H.; Kasi, A.; Baghdadi, T.A.; et al. A Phase II Basket Trial of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART SWOG 1609) in Patients with Nonpancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2020, 26, 2290–2296. [Google Scholar] [CrossRef] [Green Version]
- Wrangle, J.M.; Awad, M.M.; Badin, F.B.; Rubinstein, M.P.; Bhar, P.; Garner, C.; Reddy, S.K.; Soon-Shiong, P. Preliminary data from QUILT 3.055: A phase 2 multi-cohort study of N803 (IL-15 superagonist) in combination with checkpoint inhibitors (CPI). J. Clin. Oncol. 2021, 39, 2596. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Tredan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Kurzrock, R. Precision medicine: Lessons learned from the SHIVA trial. Lancet Oncol. 2015, 16, e579–e580. [Google Scholar] [CrossRef]
- Janku, F.; Hong, D.S.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Tsimberidou, A.M.; Stepanek, V.M.; Moulder, S.L.; Lee, J.J.; et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014, 6, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Murad, M.H.; Asi, N.; Alsawas, M.; Alahdab, F. New evidence pyramid. Evid. Based Med. 2016, 21, 125–127. [Google Scholar] [CrossRef] [Green Version]
- Bhide, A.; Shah, P.S.; Acharya, G. A simplified guide to randomized controlled trials. Acta Obs. Gynecol. Scand. 2018, 97, 380–387. [Google Scholar] [CrossRef]
- Vo, H.H.; Fu, S.; Hong, D.S.; Karp, D.D.; Piha-Paul, S.; Subbiah, V.; Janku, F.; Naing, A.; Yap, T.A.; Rodon, J.; et al. Challenges and opportunities associated with the MD Anderson IMPACT2 randomized study in precision oncology. NPJ Precis. Oncol. 2022, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Said, R.; Guibert, N.; Oxnard, G.R.; Tsimberidou, A.M. Circulating tumor DNA analysis in the era of precision oncology. Oncotarget 2020, 11, 188–211. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, M.F.; Vo, H.H.; Vining, D.; Tsimberidou, A.M. Prolonged response to treatment based on cell-free DNA analysis and molecular profiling in three patients with metastatic cancer: A case series. Ther. Adv. Med. Oncol. 2021, 13, 17588359211001538. [Google Scholar] [CrossRef]
- Chen, A.P.; Kummar, S.; Moore, N.; Rubinstein, L.V.; Zhao, Y.; Williams, P.M.; Palmisano, A.; Sims, D.; O’Sullivan Coyne, G.; Rosenberger, C.L.; et al. Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT): A Randomized Multicenter Phase II Trial. JCO Precis. Oncol. 2021, 5, 133–144. [Google Scholar] [CrossRef]
- ECOG-ACRIN Cancer Research Group. NCI-MATCH Searchable Treatment Arms Table. Available online: https://ecog-acrin.org/trials/nci-match-eay131 (accessed on 13 August 2022).
- Lih, C.J.; Harrington, R.D.; Sims, D.J.; Harper, K.N.; Bouk, C.H.; Datta, V.; Yau, J.; Singh, R.R.; Routbort, M.J.; Luthra, R.; et al. Analytical Validation of the Next-Generation Sequencing Assay for a Nationwide Signal-Finding Clinical Trial: Molecular Analysis for Therapy Choice Clinical Trial. J. Mol. Diagn. 2017, 19, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Wang, W.L.; Prieto, V.G.; Medeiros, L.J.; Kalhor, N.; Hameed, M.; Broaddus, R.; Hamilton, S.R. Validation of Immunohistochemical Assays for Integral Biomarkers in the NCI-MATCH EAY131 Clinical Trial. Clin. Cancer Res. 2018, 24, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano-Goroff, Y.R.; Drilon, A.; Stadler, Z.K. The NCI-MATCH: A National, Collaborative Precision Oncology Trial for Diverse Tumor Histologies. Cancer Cell 2021, 39, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Gray, R.; Chen, A.; Li, S.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Mitchell, E.P.; Iafrate, A.J.; Sklar, J.; et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) Trial: Lessons for Genomic Trial Design. J. Natl. Cancer Inst. 2020, 112, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.P.; O’Dwyer, P.J.; Harris, L.; Conley, B.A.; Hamilton, S.R.; Williams, M.; Gray, R.J.; Li, S.; McShane, L.M.; Rubinstein, L.V.; et al. Abstract PL03-01: NCI-MATCH: A new paradigm in the era of genomic oncology. Mol. Cancer Ther. 2018, 17, PL03-01. [Google Scholar] [CrossRef]
- Azad, N.S.; Gray, R.J.; Overman, M.J.; Schoenfeld, J.D.; Mitchell, E.P.; Zwiebel, J.A.; Sharon, E.; Streicher, H.; Li, S.; McShane, L.M.; et al. Nivolumab Is Effective in Mismatch Repair-Deficient Noncolorectal Cancers: Results from Arm Z1D-A Subprotocol of the NCI-MATCH (EAY131) Study. J. Clin. Oncol. 2020, 38, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Hong, F.; McCourt, C.; Sachdev, J.; Mitchell, E.; Zwiebel, J.; Doyle, L.; McShane, L.; Li, S.; Gray, R.; et al. AZD5363 in Patients (Pts) with Tumors with AKT Mutations: NCI-MATCH Subprotocol EAY131-Y, A Trial of the ECOG-ACRIN Cancer Research Group (EAY131-Y). Eur. J. Cancer 2018, 103, E15. [Google Scholar]
- Jhaveri, K.L.; Wang, X.V.; Makker, V.; Luoh, S.W.; Mitchell, E.P.; Zwiebel, J.A.; Sharon, E.; Gray, R.J.; Li, S.; McShane, L.M.; et al. Ado-trastuzumab emtansine (T-DM1) in patients with HER2-amplified tumors excluding breast and gastric/gastroesophageal junction (GEJ) adenocarcinomas: Results from the NCI-MATCH trial (EAY131) subprotocol Q. Ann Oncol 2019, 30, 1821–1830. [Google Scholar] [CrossRef] [Green Version]
- Krop, I.; Jegede, O.; Grilley-Olson, J.; Lauring, J.; Hamilton, S.; Zwiebel, J.; Li, S.; Rubinstein, L.; Doyle, A.; Patton, D.; et al. Results from molecular analysis for therapy choice (MATCH) arm I: Taselisib for PIK3CA-mutated tumors. J. Clin. Oncol. 2018, 36, 101. [Google Scholar] [CrossRef]
- Kummar, S.; Li, S.; Reiss, K.; Ford, J.M.; Mitchell, E.P.; Zwiebel, J.A.; Takebe, N.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Abstract CT138: NCI-MATCH EAY131 -Z1I: Phase II study of AZD1775, a wee-1 kinase inhibitor, in patients with tumors containing BRCA1 and BRCA2 mutations. Cancer Res. 2019, 79, CT138. [Google Scholar] [CrossRef]
- Chae, Y.K.; Vaklavas, C.; Cheng, H.H.; Hong, F.; Harris, L.; Mitchell, E.P.; Zwiebel, J.A.; McShane, L.; Gray, R.J.; Li, S.; et al. Molecular analysis for therapy choice (MATCH) arm W: Phase II study of AZD4547 in patients with tumors with aberrations in the FGFR pathway. J. Clin. Oncol. 2018, 36, 2503. [Google Scholar] [CrossRef]
- Kalinsky, K.; Hong, F.; McCourt, C.K.; Sachdev, J.C.; Mitchell, E.P.; Zwiebel, J.A.; Doyle, L.A.; McShane, L.M.; Li, S.; Gray, R.J.; et al. Effect of Capivasertib in Patients with an AKT1 E17K-Mutated Tumor: NCI-MATCH Subprotocol EAY131-Y Nonrandomized Trial. JAMA Oncol. 2021, 7, 271–278. [Google Scholar] [CrossRef]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, S.; Zhao, F.; Deming, D.A.; Mitchell, E.P.; Wright, J.J.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; Patton, D.R.; et al. Phase II Study of Copanlisib in Patients with Tumors with PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F. J. Clin. Oncol. 2022, 40, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cao, S.; Mashl, R.J.; Mo, C.K.; Zaccaria, S.; Wendl, M.C.; Davies, S.R.; Bailey, M.H.; Primeau, T.M.; Hoog, J.; et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidatesfor targeted treatment. Nat. Commun. 2021, 12, 5086. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Wei, Z.; Mehra, R.; Shaw, A.T.; Lieu, C.H.; Forde, P.M.; Drilon, A.E.; Mitchell, E.P.; Wright, J.J.; Takebe, N.; et al. Crizotinib in patients with tumors harboring ALK or ROS1 rearrangements in the NCI-MATCH trial. NPJ Precis. Oncol. 2022, 6, 13. [Google Scholar] [CrossRef]
- Johnson, D.B.; Zhao, F.; Noel, M.; Riely, G.J.; Mitchell, E.P.; Wright, J.J.; Chen, H.X.; Gray, R.J.; Li, S.; McShane, L.M.; et al. Trametinib Activity in Patients with Solid Tumors and Lymphomas Harboring BRAF Non-V600 Mutations or Fusions: Results from NCI-MATCH (EAY131). Clin. Cancer Res. 2020, 26, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II Study of AZD4547 in Patients with Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J. Clin. Oncol. 2020, 38, 2407–2417. [Google Scholar] [CrossRef]
- Cleary, J.M.; Wang, V.; Heist, R.S.; Kopetz, E.S.; Mitchell, E.P.; Zwiebel, J.A.; Kapner, K.S.; Chen, H.X.; Li, S.; Gray, R.J.; et al. Differential Outcomes in Codon 12/13 and Codon 61 NRAS-Mutated Cancers in the Phase II NCI-MATCH Trial of Binimetinib in Patients with NRAS-Mutated Tumors. Clin. Cancer Res. 2021, 27, 2996–3004. [Google Scholar] [CrossRef]
- Krop, I.E.; Jegede, O.A.; Grilley-Olson, J.E.; Lauring, J.D.; Mitchell, E.P.; Zwiebel, J.A.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; et al. Phase II Study of Taselisib in PIK3CA-Mutated Solid Tumors Other Than Breast and Squamous Lung Cancer: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol I. JCO Precis. Oncol. 2022, 6, e2100424. [Google Scholar] [CrossRef]
- Bedard, P.L.; Li, S.; Wisinski, K.B.; Yang, E.S.; Limaye, S.A.; Mitchell, E.P.; Zwiebel, J.A.; Moscow, J.A.; Gray, R.J.; Wang, V.; et al. Phase II Study of Afatinib in Patients with Tumors with Human Epidermal Growth Factor Receptor 2-Activating Mutations: Results from the National Cancer Institute-Molecular Analysis for Therapy Choice ECOG-ACRIN Trial (EAY131) Subprotocol EAY131-B. JCO Precis. Oncol. 2022, 6, e2200165. [Google Scholar] [CrossRef]
- Pisick, E.P.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Yang, E.S.-H.; Dib, E.G.; Burgess, E.F.; Zakem, M.H.; Rohatgi, N.; Bilen, M.A.; et al. Olaparib (O) in patients (pts) with prostate cancer with BRCA1/2 inactivating mutations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 2020, 38, 5567. [Google Scholar] [CrossRef]
- Ahn, E.R.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Calfa, C.J.; Alva, A.S.; Suhag, V.S.; Hamid, O.; Dotan, E.; Yang, E.S.-H.; et al. Olaparib (O) in patients (pts) with pancreatic cancer with BRCA1/2 inactivating mutations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 2020, 38, 4637. [Google Scholar] [CrossRef]
- Mileham, K.F.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Yang, E.S.; Alese, O.B.; Jain, A.; Duvivier, H.L.; Palmbos, P.; Ahn, E.R.; et al. Olaparib (O) in patients (pts) with solid tumors with ATM mutation or deletion: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Cancer Res. 2022, 82 (Suppl. 12), CT110. [Google Scholar] [CrossRef]
- Ahn, E.R.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Al Baghdadi, T.; Baron, A.D.; Krauss, J.C.; Balmanoukian, A.S.; Bauman, J.R.; Hameed, M.K.; et al. FPN 98P: Olaparib in Patients with Solid Tumors with BRCA1/2 Mutation: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Available online: https://www.asco.org/sites/new-www.asco.org/files/content-files/ESMO-2022-Olaparib-Colorectal-ATM-TAPUR-Poster.pdf (accessed on 9 August 2022).
- Pisick, E.P.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, L.; Worden, F.P.; Bauman, J.R.; Fu, S.; Leidner, R.S.; Balmanoukian, A.S.; Calfa, C.; et al. Palbociclib (P) in patients (pts) with head and neck cancer (HNC) with CDKN2A loss or mutation: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 2021, 39, 6043. [Google Scholar] [CrossRef]
- Ahn, E.R.; Mangat, P.K.; Garrett-Mayer, E.; Halabi, S.; Dib, E.G.; Haggstrom, D.E.; Alguire, K.B.; Calfa, C.J.; Cannon, T.L.; Crilley, P.A.; et al. Palbociclib in Patients with Non–Small-Cell Lung Cancer with CDKN2A Alterations: Results from the Targeted Agent and Profiling Utilization Registry Study. JCO Precis. Oncol. 2020, 4, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Schuetze, S.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, L.; Meric-Bernstam, F.; Farrington, L.C.; Calfa, C.; D’Andre, S.D.; Livingston, M.B.; Thota, R.; et al. Palbociclib (P) in patients (pts) with soft tissue sarcoma (STS) with CDK4 amplification: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 2021, 39, 11565. [Google Scholar] [CrossRef]
- Gupta, R.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; D’Andre, S.D.; Meiri, E.; Shrestha, S.; Warren, S.L.; Ranasinghe, S.; Schilsky, R.L. Pertuzumab plus trastuzumab (P+T) in patients (Pts) with colorectal cancer (CRC) with ERBB2 amplification or overexpression: Results from the TAPUR Study. J. Clin. Oncol. 2020, 38, 132. [Google Scholar] [CrossRef]
- Ali-Ahmad, H.M.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, L.; Ahn, E.; Chan, J.; Maitland, M.L.; Balmanoukian, A.S.; Patel, S.R.; Reese, Z.; et al. Pertuzumab plus trastuzumab (P+T) in patients (Pts) with uterine cancer (UC) with ERBB2 or ERBB3 amplification, overexpression or mutation: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 2021, 39, 5508. [Google Scholar] [CrossRef]
- Ganti, A.K.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Dib, E.G.; Duvivier, H.; Ahn, E.; Behl, D.; Borghaei, H.; Balmanoukian, A.S.; et al. FPN 978P: Pertuzumab Plus Trastuzumab in Patients with Lung Cancer with ERBB2 Mutation or Amplification: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. 2022. Available online: https://www.researchgate.net/publication/363523507_978P_Pertuzumab_plus_trastuzumab_PT_in_patients_pts_with_lung_cancer_LC_with_ERBB2_mutation_mut_or_amplification_amp_Results_from_the_Targeted_Agent_and_Profiling_Utilization_Registry_TAPUR_study (accessed on 19 March 2023).
- Alva, A.S.; Mangat, P.K.; Garrett-Mayer, E.; Halabi, S.; Hansra, D.; Calfa, C.J.; Khalil, M.F.; Ahn, E.R.; Cannon, T.L.; Crilley, P.; et al. Pembrolizumab in Patients with Metastatic Breast Cancer with High Tumor Mutational Burden: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. J. Clin. Oncol. 2021, 39, 2443–2451. [Google Scholar] [CrossRef]
- Meiri, E.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Shrestha, S.; Ahn, E.R.; Osayameh, O.; Perla, V.; Schilsky, R.L. Pembrolizumab (P) in patients (Pts) with colorectal cancer (CRC) with high tumor mutational burden (HTMB): Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. J. Clin. Oncol. 2020, 38, 133. [Google Scholar] [CrossRef]
- Calfa, C.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Ahn, E.; Gogineni, K.; Rohatgi, N.; Burness, M.L.; Gaba, A.; Hamid, O.; et al. Abstract CT173: Sunitinib (S) in patients (pts) with metastatic breast cancer (mBC) with FGFR1 mutations or amplifications: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Cancer Res. 2021, 81, CT173. [Google Scholar] [CrossRef]
- Klute, K.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Nazemzadeh, R.; Yost, K.J.; Butler, N.L.; Perla, V.; Schilsky, R.L. Cobimetinib plus vemurafenib (C+V) in patients (Pts) with colorectal cancer (CRC) with BRAF V600E mutations: Results from the TAPUR Study. J. Clin. Oncol. 2020, 38, 122. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Rothe, M.; Garrett-Mayer, E.; Gutierrez, R.; Ahn, E.R.; Cannon, T.L.; Powell, S.F.; Krauss, J.C.; Reynolds, C.M.; Mehren, M.V.; et al. Cobimetinib plus vemurafenib (C+V) in patients (Pts) with solid tumors with BRAF V600E/d/k/R mutation: Results from the targeted agent and profiling utilization registry (TAPUR) study. J. Clin. Oncol. 2022, 40, 3008. [Google Scholar] [CrossRef]
- Baghdadi, T.A.; Halabi, S.; Garrett-Mayer, E.; Mangat, P.K.; Ahn, E.R.; Sahai, V.; Alvarez, R.H.; Kim, E.S.; Yost, K.J.; Rygiel, A.L.; et al. Palbociclib in Patients with Pancreatic and Biliary Cancer with CDKN2A Alterations: Results from the Targeted Agent and Profiling Utilization Registry Study. JCO Precis. Oncol. 2019, 1, 1–8. [Google Scholar] [CrossRef]
- Al Baghdadi, T.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Rich, P.; Ahn, E.R.; Chai, S.; Rygiel, A.L.; Osayameh, O.; Antonelli, K.R.; et al. Sunitinib in Patients with Metastatic Colorectal Cancer (mCRC) with FLT-3 Amplification: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 743–750. [Google Scholar] [CrossRef]
- Fisher, J.G.; Tait, D.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Schink, J.C.; Alvarez, R.H.; Veljovich, D.; Cannon, T.L.; Crilley, P.A.; et al. Cetuximab in Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Ovarian Cancer without KRAS, NRAS, or BRAF Mutations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 733–741. [Google Scholar] [CrossRef]
- Vaccaro, G.M.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Hwang, J.J.; Alese, O.B.; Khalil, M.F.; Hameed, M.K.; Duvivier, H.L.; Cannon, T.L.; et al. Abstract 107: Nivolumab + Ipilimumab in Patients with Colorectal Cancer with High Tumor Mutational Burden (hTMB): Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Available online: https://www.asco.org/sites/new-www.asco.org/files/content-files/about-asco/Nivo-Ipi-CRC-HTMB-2022-GIS-Poster.pdf (accessed on 13 August 2022).
- Grem, J.L.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Khalil, M.F.; Salmon, J.S.; Rogosin, S.O.; Cannon, T.L.; O’Lone, R.; Grantham, G.N.; et al. Abstract 106: Temsirolimus in Patients with Colorectal Cancer with PIK3CA Mutation: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Available online: https://www.asco.org/sites/new-www.asco.org/files/content-files/about-asco/Temsirolimus-CRC-PIK3CA-2022-GIS-Poster.pdf (accessed on 13 August 2022).
- Srkalovic, G.; Rothe, M.; Mangat, P.K.; Garrett-Mayer, E.; Nazemzadeh, R.; Cannon, T.L.; Duvivier, H.L.; Yost, K.J.; Pakkala, S.; Alva, A.S.; et al. Abstract 3114: Temsirolimus in Patients with Solid Tumors with mTOR Mutation: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Available online: https://www.asco.org/sites/new-www.asco.org/files/content-files/Temsirolimus-Collapsed-mTOR-Poster-ASCO-2022.pdf (accessed on 9 August 2022).
- ASCOTAPUR. Targeted Agent and Profiling Utilization Registry (TAPURTM) Study. Available online: https://www.tapur.org/ (accessed on 30 July 2020).
- JCO Precision Oncology. Summary of Cohort Activity in the TAPUR Study. Available online: https://www.asco.org/research-data/tapur-study/study-results (accessed on 9 August 2022).
- Hoes, L.R.; van Berge Henegouwen, J.M.; van der Wijngaart, H.; Zeverijn, L.J.; van der Velden, D.L.; van de Haar, J.; Roepman, P.; de Leng, W.J.; Jansen, A.M.L.; van Werkhoven, E.; et al. Patients with Rare Cancers in the Drug Rediscovery Protocol (DRUP) Benefit from Genomics-Guided Treatment. Clin. Cancer Res. 2022, 28, 1402–1411. [Google Scholar] [CrossRef]
- Adams, S.; Othus, M.; Patel, S.P.; Miller, K.D.; Chugh, R.; Schuetze, S.M.; Chamberlin, M.D.; Haley, B.J.; Storniolo, A.M.V.; Reddy, M.P.; et al. A Multicenter Phase II Trial of Ipilimumab and Nivolumab in Unresectable or Metastatic Metaplastic Breast Cancer: Cohort 36 of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART, SWOG S1609). Clin. Cancer Res. 2022, 28, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Ford, J.M.; O’Dwyer, P.J.; Shapiro, G.I.; McShane, L.M.; Freidlin, B.; O’Cearbhaill, R.E.; George, S.; Glade-Bender, J.; Lyman, G.H.; et al. National Cancer Institute Combination Therapy Platform Trial with Molecular Analysis for Therapy Choice (ComboMATCH). Clin. Cancer Res. 2023, OF1–OF11. [Google Scholar] [CrossRef]
- Little, R.F.; Othus, M.; Assouline, S.; Ansher, S.; Atallah, E.L.; Lindsley, R.C.; Freidlin, B.; Gore, S.D.; Harris, L.; Hourigan, C.S.; et al. Umbrella Trial in Myeloid Malignancies: The Myelomatch National Clinical Trials Network Precision Medicine Initiative. Blood 2022, 140, 9057–9060. [Google Scholar] [CrossRef]
- SWOG Cancer Research Network. iMATCH Pilot: Immunotherapy Master Protocol. Available online: https://www.swog.org/news-events/news/2022/04/22/imatch-pilot-immunotherapy-master-protocol (accessed on 10 March 2023).
- U.S. FOOD & DRUG. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 10 March 2023).



{kind=link}
{kind=link}
| First, Last Author, Year | Treatment | Cancer Type | Molecular Alteration(s) | Enrollment (N) | Safety | Clinical Outcomes |
|---|---|---|---|---|---|---|
| Pilot study using tumor molecular profiling (MP) to identify targets and matched treatments for refractory cancer | ||||||
| Von Hoff; Penny, 2010 [10] | Various treatments * | Diverse solid tumors | Oligonucleo-tide microarray (MA) gene expression assays | 106 | TRAE, n = 9 (anemia, n = 2; neutropenia, n = 2; dehydration, n = 1; pancreatitis, n = 1; nausea, n = 1; vomiting, n = 1; febrile neutropenia, n = 1) Treatment discontinuation, n = 1 (patient’s request, grade 2 fatigue). | Molecular target detected, 98% (84/86 had MP attempted); 66 of 84 patients were treated according to MP results; 18 (27%) of 66 patients had a PFS ratio (PFS on MP-selected therapy/PFS on prior therapy) of ≥1.3. |
| IMPACT (Initiative for Molecular Profiling and Advanced Cancer Therapy) | ||||||
| Tsimberidou; Kurzrock, 2012 [9] | Matched versus unmatched therapy following NGS results | Diverse solid tumors | NGS | 1144 | N/A | ORR, matched vs. unmatched, 27% vs. 5% (p < 0.0001) mTTF, 5.2 vs. 2.2 months (p < 0.0001) mOS, 13.4 vs. 9.0 months (p = 0.017) |
| Tsimberidou; Berry, 2014 [11] | Matched versus unmatched therapy following NGS results | Diverse solid tumors | NGS | 1542 | N/A | ORR, matched vs unmatched, 12% vs. 5% (p < 0.0001) mPFS, 3.9 vs. 2.2 months (p = 0.001) mOS, 11.4 vs. 8.6 months (p = 0.04) Matched therapy independent factor for response and PFS mOS (2-month Landmark analysis): Responders vs. non-responders, 30.5 months vs 11.3 months (p = 0.01) |
| Tsimberidou; Kurzrock, 2017 [12] | Matched versus unmatched therapy following NGS results | Diverse solid tumors | NGS | 1436 | N/A | MTT (n = 390), non-MTT (n = 247) MTT vs. non-MTT, ORR (PR): 11% vs. 5% (p = 0.0099) FFS, 3.4 vs. 2.9 months (p = 0.0015) OS, 8.4 vs. 7.3 months (p = 0.041) |
| Tsimberidou; Kurzrock, 2019 [13] | Matched versus unmatched therapy following NGS results | Diverse solid tumors | NGS | 3487 | N/A | ORR, MTT 16.4%, non-MTT 5.4% (p < 0.0001) SD ≥ 6 months, MTT 35.3%, non-MTT 20.3%, (p < 0.001) mPFS, MTT 4.0, non-MTT 2.8 months (p < 0.0001) MTT vs. non-MTT: mOS, 9.3 months vs. 7.3 months; 3-yr OS, 15% vs. 7%; 10-yr OS, 6% vs. 1% (p < 0.0001) |
| Investigation of Profile-Related Evidence Determining Individualized Cancer Therapy (I-PREDICT) | ||||||
| Schwaederle; Kurzrock, 2016 [14] | Matched versus unmatched therapy following NGS results | Metastatic/ refractory, therapy-naive solid tumors | PD-L1, ctDNA, TMB, MSI | 347 | N/A | DCR, matched, 34.5% vs. unmatched therapy, 16.1% (p ≤ 0.005); mPFS, months: matched 4.0 vs. unmatched therapy, 3.0 (p = 0.039); mOS, months: matched, 15.7 (matching score > 0.2) vs. unmatched, 10.6 months (matching-score of ≤ 0.2) (p = 0.040) |
| Worldwide Innovative Network trial with genomics and transcriptomics (WINTHER) | ||||||
| Rodon; Kurzrock, 2019 [15] | Matched versus unmatched therapy following MP results | Diverse advanced metastatic solid tumors | NGS and transcriptomics | 303 | High-grade AEs: diarrhea, rash, fatigue/weakness | Evaluable for treatment, n = 107 (35%; arm A, n = 69; arm B, n = 38). SD ≥ 6 months/PR/CR, 26.2% (arm A: 23.2%; arm B: 31.6% (p = 0.37)). PFS ratio (patient proportion with WINTHER vs. previous therapy of >1.5) = 22.4% |
| Dual Anti-CTLA-4 and Anti-PD-1 blockade in Rare Tumors (DART) | ||||||
| Patel; Kurzrock, 2020 [16] | Ipilimumab + nivolumab | Non-pancreatic neuro-endocrine carcinoma | Dual anti-CTLA-4 and anti-PD-1 | 32 Most common primary sites: GI (47%; N = 15); lung (19%; N = 6) | G3/4 AEs: hypothyroidism (31%), fatigue (28%), ALT elevation (9%) | SD, 41% (6%, SD ≥ 6 months) ORR, 25% (CR, 3%, n = 1; PR, 22%, n = 7) High-grade: ORR, 44% Low/intermediate-grade: ORR, 0% 6-month PFS: 31% Evaluable for OS, N = 18 Median OS: 11 months (95 CI, 6-NR) |
| Octopus study, phase I/II | ||||||
| M. Wrangle; Soon-Shiong, 2021 [17] | Quilt-3.055 (NCT03228667) N803 (IL-15 superagonist) plus investigator choice † | Diverse solid tumors | T-cell modulation, PD-L1 | 135 (60% NSCLC) | G1-2 TRAE: injection site reaction 68%, chills 32%, fatigue 26%, pyrexia 26%, flu-like illness 14%, nausea 12% | Response: non-evaluable, 12% CR 0%, PR 8%, SD 51%, PD 29% PFS, median, 3.9 months OS, median, 13.8 months |
| Published Data (First, Last Author, Year) | Treatment | Cancer Type | Molecular Alteration(s) | Enrollment (N) | Safety | Clinical Outcomes |
|---|---|---|---|---|---|---|
| NCI-MATCH (as of 13 August 2022 update, 39 arms: 2 open, 12 published, 10 presented, 15 closed) | ||||||
| Azad; Flaherty, 2020 [35] | Nivolumab | Non-CRCs | Mismatch repair-deficient | 42 | G4 toxicities, n = 3 (sepsis, n = 2) G1-3 AEs: fatigue (40%), anemia (33%), rash (17%), hypoalbuminemia (17%) | ORR, 36%; SD, 21%; 6-month PFS, 51.3%; 12-month PFS, 46.2%; 18-month PFS, 31.4%; Median OS, 17.3 months |
| Kalinsky; Flaherty, 2021 [41] | Capivasertib | Diverse tumor types | AKT1 E17K mutations | 35 | Discontinued: AEs, 31% (11/35); G3 treatment-related AE: hyperglycemia (n = 8, 23%) and rash (n = 4, 11%); G4 hyperglycemia (n = 1) | ORR, 28.6%; 6-month PFS, 50%; Median OS, 14.5 months |
| Salama; Flaherty, 2020 [42] | Dabrafenib and trametinib | Diverse tumor types | BRAF V600E mutations | 35 | G3 AEs: fatigue (n = 4); neutropenia (n = 3); hyponatremia (n = 2); G4 sepsis (n = 1). | ORR, 38%; Median PFS, 11.4 months; Median OS, 28.6 months |
| Damodaran; Flaherty, 2022 [43] | Copanlisib | Diverse tumor types | PIK3CA mutations | 35 (25 were included in the primary efficacy analysis as prespecified in the protocol) | G3 AEs: hyperglycemia (n = 7); rash, maculopapular (n = 2); mucositis, oral (n = 1); vomiting (n = 1); weight loss (n = 1); general muscle weakness (n = 2); pruritus (n = 1); hypertension (n = 9); dehydration (n = 2); acute kidney injury (n = 1); dizziness (n = 1); hypophosphatemia (n = 1); hypoglycemia (n = 1); hypoxia (n = 1); meningitis (n = 1); oral pain (n = 1); syncope (n = 1). G4 AE: hyperglycemia (n = 1) | ORR, 16%; Median PFS, 3.4 months; median OS, 5.9 months |
| Mansfield; Flaherty, 2022 [45] | Crizotinib | Diverse tumor types | ALK or ROS1 rearrangements | 9 (5 ALK, 4 ROS1) | ALK: G3 AEs: AST increased (n = 1); hypophosphatemia (n = 1) G4 AEs: ALT increased (n = 1); AST increased (n = 1); blood bilirubin increased (n = 1); hyponatremia (n = 1) ROS1: G3 AEs: abdominal pain (n = 1); ALC decreased (n = 1); acute kidney injury (n = 1) | ALK: ORR, 50%; median PFS, 3.8 months; median OS, 4.3 months ROS1: ORR, 25%; median PFS, 4.3 months; median OS, 6.2 months |
| TAPUR https://www.asco.org/research-data/tapur-study/study-results, accessed on 9 August 2022, cohort updated 8 April 2022 | ||||||
| Pisick; Schilsky, Meeting Abstract, 2020 [51] | Olaparib | Prostate cancer | BRCA1/2 inactivating mutations | 29 (25 included in efficacy analyses) | G3-4 AE (n = 6): anemia, aspiration, dehydration, diabetic ketoacidosis, fatigue, and neutropenia | DCR, 68%; ORR, 36%; median PFS, 41.0 weeks; median OS, 75.4 weeks; 1-year OS rate, 79.4% |
| Ahn; Schilsky, Meeting Abstract, 2020 [52] | Olaparib | Pancreatic cancer | BRCA1/2 inactivating mutations | 30 (28 included in efficacy analyses) | G3-4 AE (n = 7): anemia, diarrhea, fever, elevated liver enzymes, enterocolitis, increased bilirubin, oral mucositis | DCR, 31%; ORR, 4%; median PFS, 8.1 weeks; median OS, 43 weeks; 1-year OS rate, 47.2% |
| Mileham; Schilsky Meeting Abstract, 2022 [53] | Olaparib | Diverse tumor types | ATM mutations or deletions | 39 (37 included in efficacy analyses) | G3-4 AE or SAE (n = 9): anemia, anorexia, colitis, dehydration, dizziness, fatigue, hypokalemia, lung infection, nausea, proteinuria, urinary tract infection, urinary tract obstruction | DCR, 27%; ORR, 8%; median PFS, 8.6 weeks; median OS, 40.9 weeks |
| Ahn; Schilsky, Meeting Abstract, 2022 [54] | Olaparib | Diverse tumor types | Germline or somatic BRCA1/BRCA2 inactivating mutations | 32 (32 included in efficacy analyses) | G3-4 AE or SAE (n = 12): anemia, dyspnea, fatigue, fever, generalized muscle weakness, tumor lysis syndrome, leukopenia/thrombocytopenia | DCR, 41%; ORR, 25%; median PFS, 15.7 weeks; median OS, 45 weeks |
| Pisick; Schilsky, Meeting Abstract, 2021 [55] | Palbociclib | Head and neck cancer | CDKN2A loss or mutation | 28 (28 included in efficacy analyses) | G3-4 AEs (n = 13): Cytopenias, hypocalcemia, syncope G5 AEs (n = 1): respiratory failure | ORR, 0%; DCR, 37%; median PFS, 9.4 weeks; median OS, 42.0 weeks |
| Ahn; Schilsky, 2020 [56] | Palbociclib | Non-small cell lung cancer | CDKN2A alterations | 29 (27 included in efficacy analyses) | G3-4 AE or SAE (n = 11): most common, cytopenias | DCR, 31%; median PFS, 8.1 weeks; median OS, 21.6 weeks |
| Schuetze; Schilsky, Meeting Abstract, 2021 [57] | Palbociclib | Soft tissue sarcoma | CDK4 amplification | 29 (28 included in efficacy analyses) | G3-4 AEs (n = 14): most common, leukopenia/thrombocytopenia | DCR, 48%; ORR, 3.7%; median PFS, 16.1 weeks; median OS, 68.7 weeks; 1-year OS rate, 53.6% |
| Gupta; Schilsky, Meeting Abstract, 2020 [58] | Pertuzumab and trastuzumab | CRC | ERBB2 amplification or overexpression | 28 (28 included in efficacy analyses) | G3 SAE: anemia, infusion reaction, left ventricular dysfunction | DCR, 50%; ORR, 14%; median PFS, 17.2 weeks; 1-year OS rate, 58% |
| Ali-Ahmad; Schilsky, Meeting Abstract, 2021 [59] | Pertuzumab and trastuzumab | Uterine cancer | ERBB2 or ERBB3 amplification, overexpression, or mutation | 28 (28 included in efficacy analyses) | G3 AE (n = 1): muscle weakness | DCR, 37%; ORR, 7.1%; median OS, 28.1 weeks; 1-year OS rate, 53.4% |
| Gant; Schilsky, Meeting Abstract, 2022 [60] | Pertuzumab and trastuzumab | Bronchus and lung | ERBB2/ERBB3 amplification, mutation, or overexpression | 28 (28 included in efficacy analyses) | G3-4 AE or SAE (n = 5): ALT increased, AST increased, dyspnea, fatigue, infusion-related reaction, nausea, vomiting | DCR, 37%; ORR, 11%; Median OS, 54.4 weeks |
| Alva; Schilsky, 2021 [61] | Pembrolizumab | Metastatic breast cancer | High tumor mutational burden | 28 (28 included in efficacy analyses) | G3 (n = 4): pulmonary embolism, weight loss, hypoalbuminemia, hyponatremia G2-3 SAE (n = 4): colonic obstruction, diarrhea, urinary tract infection, hepatic failure | DCR, 37%; ORR, 21%; median PFS,10.6 weeks; median OS, 30.6 weeks |
| Meiri; Schilsky, Meeting Abstract, 2020 [62] | Pembrolizumab | CRC | High tumor mutational burden | 28 (27 included in efficacy analyses) | G3 AE (n = 2, each): abdominal infection, anorexia, colitis, diarrhea, fatigue, nausea, vomiting G3 SAE (n = 1): acute kidney injury | DCR, 28%; ORR, 4%; median PFS, 9.3 weeks; 1-year OS rate, 45.6% |
| Calfa; Schilsky, Meeting Abstract, 2021 [63] | Sunitinib | Metastatic breast cancer | FGFR1 alterations | 30 (27 included in efficacy analyses) | G3 AEs (n = 9): cytopenia, encephalopathy, febrile neutropenia, increased alkaline phosphatase, palmar-plantar erythrodysesthesia syndrome, vomiting G4 AEs (n = 2): cytopenia, hypertension | DCR, 29%; ORR, 7%; median PFS, 8.7 weeks; median OS, 33.9 weeks |
| Klute; Schilsky, Meeting Abstract, 2020 [64] | Cobimetinib and vemurafenib | CRC | BRAF V600E mutations | 30 (28 included in efficacy analyses) | G3 AE/SAE (n = 12): elevated liver enzymes, decreased lymphocytes, dyspnea, diarrhea, fatigue, hypercalcemia, hypophosphatemia, rash, photosensitivity, upper GI hemorrhage, vomiting | DCR, 57%; ORR, 29%; median PFS, 15.8 weeks; median OS, 38.9 weeks |
| Meric-Bernstam; Schilsky, Meeting Abstract, 2022 [65] | Cobimetinib and vemurafenib | Diverse tumor types | BRAF_V600E/D/K/R mutation | 31 (28 included in efficacy analyses) | G3 AE (n = 17): rash, anemia, hypokalemia, increased ALP, increased AST, increased ALT, increased CPK, diarrhea, increased GGT, hypophosphatemia, decreased ALC, multiple SCCs of skin, decreased platelet count, treatment-related secondary malignancy G4 AE (n = 1): increased GGT | DCR, 68%; ORR, 57%; median PFS, 5.8 months; median OS, 15.2 months |
| NCI-MATCH | ||||||
|---|---|---|---|---|---|---|
| First, Last Author, Year | Treatment | Cancer Type | Molecular Alteration(s) | Enrollment (N) | Safety | Clinical Outcomes |
| Chae; Flaherty, 2020 [47] | AZD4547 (FGFR inhibitor) | Diverse tumor types | FGFR pathway aberrations | 70 (48 eligible and treated) | G3 AEs: oral mucositis (n = 7); constipation (n = 1); dry eye (n = 1); anemia (n = 2); palmar-plantar erythrodysesthesia (n = 3); peripheral sensory neuropathy (n = 1); dizziness (n = 1); abdominal pain (n = 1); esophageal pain (n = 1); small intestinal obstruction (n = 1); laryngeal mucositis (n = 1); syncope (n = 1); febrile neutropenia (n = 1); increased ALP (n = 1); increased ALT (n = 3); increased AST (n = 4); hypernatremia (n = 1); hypophosphatemia (n = 1); decreased neutrophil count (n = 1); increased GGT (n = 1) G4 AEs: diarrhea (n = 1), sepsis (n = 1) | 6-month PFS, 15%; median PFS, 3.4 months |
| Johnson; Flaherty, 2020 [46] | Trametinib | Solid tumors and lymphomas | BRAF non-V600 mutations or fusions | 50 (32 eligible and treated) | G3 AEs: anemia (n = 5); nausea (n = 1); vomiting (n = 1); anorexia (n = 1); hypoalbuminemia (n = 1); pruritus (n = 1); rash acneiform (n = 1); rash maculopapular (n = 1); other skin and subcutaneous tissue disorders (n = 1) | 6-month PFS, 17%; median PFS, 1.8 months; 6-month OS, 46%; median OS, 5.7 months |
| Jhaveri; Flaherty, 2019 [37] | Ado-trastuzu-mab emtansine (T-DM1) | Diverse tumor types other than breast and gastroeso-phageal tumors | HER2 amplification at a copy number >7 | 38 (36 included in efficacy analysis) | G3 AEs: anemia (n = 3); fatigue (n = 2); fever (n = 1); nausea (n = 1); ileal obstruction (n = 1); ALP increase (n = 1); AST increase (n = 1); lymphocyte count decrease (n = 1); neutrophil count decrease (n = 1); platelet count decrease (n = 2); anorexia (n = 2); epistaxis (n = 1); hypoxia (n = 1); muscle weakness, lower limb (n = 1); dehydration (n = 1); investigations, other (n = 1); urinary tract infection (n = 1); upper respiratory infection (n = 1); diarrhea (n = 1); blurred vision (n = 1) | 6-month PFS, 23.3%; median OS, 8.4 months; ORR, 5.6% |
| Bedard; Flaherty, 2022 [50] | Afatinib | Diverse tumor types | ERBB2-activating mutations | 59 (40 enrolled, 37 included in efficacy analysis) | G3 AEs: diarrhea (18.9%), mucositis (8.1%), and fatigue (8.1%) | 6-month PFS, 12.0%; median PFS, 1.7 months; median OS, 6.5 months; ORR, 2.7% |
| Cleary; Flaherty, 2021 [48] | Binimetinib | Diverse tumor types (melanoma excluded) | Codon 12/13 and codon 61 NRAS-mutated | 53 (47 eligible and included in efficacy analysis) | G3 AEs: heart failure (n = 1); myocardial infarction (n = 1); eye disorders (n = 1); mucositis oral (n = 1); nausea (n = 1); small intestinal obstruction (n = 1); fatigue (n = 1); edema limbs (n = 1); urinary tract infection (n = 1); ALT increased (n = 1); ALP increased (n = 1); AST increased (n = 1); CPK increased (n = 2); lymphocyte count decreased (n = 2); white blood cell decreased (n = 1); ejection fraction decreased (n = 1); anorexia (n = 1); dehydration (n = 1); hypoalbuminemia (n = 1); hyponatremia (n = 1); hypophosphatemia (n = 1); muscle weakness, lower limb (n = 1); muscle weakness, upper limb (n = 1); syncope (n = 1); rash acneiform (n = 1); skin and subcutaneous tissue disorders (n = 1); hypertension (n = 6) G5 AE: multi-organ failure (n = 1) | 6-month PFS, 29.2%; median PFS, 3.5 months; median OS, 10.5 months; ORR, 2.1% |
| Krop; Flaherty, 2022 [49] | Taselisib | Solid tumors other than breast and squamous cell lung cancer | PIK3CA mutations | 70 (61 eligible and initiated protocol) | G3 AEs: diarrhea (n = 7); fatigue (n = 1); nausea (n = 2); hyperglycemia (n = 2); anorexia (n = 1); mucositis, oral (n = 1); AST increased (n = 1); abdominal pain (n = 1); vomiting (n = 1); hypokalemia (n = 1); hyponatremia (n = 3); dehydration (n = 2); hypertension (n = 1); weight loss (n = 1); lung infection (n = 3); pneumonitis (n = 1); thromboembolic event (n = 1); adult respiratory distress syndrome (n = 1); blood bilirubin increased (n = 1); dysphagia (n = 1) G4 AEs: hyperglycemia (n = 1) G5 AEs: neoplasms benign, malignant, and unspecified (n = 1); sudden death NOS (n = 1) | 6-month PFS, 19.9%; median PFS, 3.1 months; 6-month OS, 60.7%; median OS, 7.2 months |
| TAPUR | ||||||
| Baghdadi; Schilsky 2019 [66] | Palbociclib | Pancreatic | CDKN2A loss or mutation | 12 (10 evaluable patients) | ≥G3 AEs (n = 1): fatigue | No patients had objective response or stable disease at 16 weeks. Median PFS, 7.2 weeks; median OS, 12.4 weeks |
| Baghdadi; Schilsky 2019 [66] | Palbociclib | Biliary cancers | CDKN2A loss or mutation | 10 (10 evaluable patients) | G3 (n = 1): muscle weakness and port infection; G4 (n = 4): thrombocytopenia | No patients had objective response or stable disease at 16 weeks. Median PFS, 7.3 weeks; median OS, 11.1 weeks |
| Baghdadi; Schilsky 2020 [67] | Sunitinib | Metastatic CRC | FLT-3 amplification | 10 (10 evaluable patients) | G3 AEs (n = 1): diarrhea | Median PFS, 10.1 weeks; median OS, 38 weeks |
| Fisher; Schilsky, 2020 [68] | Cetuximab | Advanced breast, NSCLC, and ovarian cancer | KRAS, NRAS, BRAF mutations | 49 (28 evaluable patients) | ≥G3 AEs (n = 6) BC: hypomagnesemia; NSCLC: anemia, hyponatremia, hypophosphatemia, hypokalemia, hypomagnesemia, and cytopenia; OC: fever, infusion-related reaction, hypotension, nausea, vomiting | BC: Median PFS, 6.7 weeks; median OS, 14.1 weeks NSCLC: Median PFS, 8 weeks; median OS, 22.7 weeks OC: Median PFS, 8 weeks; median OS, 21.6 weeks |
| Vaccaro; Schilsky, Meeting Abstract, 2019 [69] | Nivolumab and ipilimumab | CRC | High tumor mutational burden | 12 (10 included in efficacy analyses) | G3-4 AEs (n = 4): myasthenia gravis, diarrhea, glucose intolerance, hyperglycemia, small intestinal obstruction | DCR, 10%; ORR, 10%; median PFS, 8.9 weeks; median OS, 42.9 weeks |
| Grem; Schilsky, Meeting Abstract, 2022 [70] | Temsiro-limus | CRC | PIK3CA mutation | 10 (10 included in efficacy analyses) | G3-4 AEs (n = 6): acute kidney injury, dehydration, thrombocytopenia, hypertriglyceridemia, mucositis, neutropenia, scrotal and penile edema | DCR, 10%; ORR, 0%; median PFS, 8.1 weeks; median OS, 38.7 weeks |
| Srkalovic; Schilsky, Meeting Abstract, 2022 [71] | Temsiro-limus | Diverse cancer types | mTOR mutation/ amplification | 29 (20 included in efficacy analyses) | G3-4 AEs (n = 8): acute kidney injury, epistaxis, hyperglycemia, hypertension, hypertriglyceridemia, oral mucositis, leukopenia, thrombocytopenia, pneumonitis | DCR, 45%; ORR, 10%; median PFS, 16.1 weeks; median OS, 48.7 weeks |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, I.-W.; Vo, H.H.; Chen, Y.-S.; Baysal, M.A.; Kahle, M.; Johnson, A.; Tsimberidou, A.M. Precision Oncology: Evolving Clinical Trials across Tumor Types. Cancers 2023, 15, 1967. https://doi.org/10.3390/cancers15071967
Song I-W, Vo HH, Chen Y-S, Baysal MA, Kahle M, Johnson A, Tsimberidou AM. Precision Oncology: Evolving Clinical Trials across Tumor Types. Cancers. 2023; 15(7):1967. https://doi.org/10.3390/cancers15071967
Chicago/Turabian StyleSong, I-Wen, Henry Hiep Vo, Ying-Shiuan Chen, Mehmet A. Baysal, Michael Kahle, Amber Johnson, and Apostolia M. Tsimberidou. 2023. "Precision Oncology: Evolving Clinical Trials across Tumor Types" Cancers 15, no. 7: 1967. https://doi.org/10.3390/cancers15071967
APA StyleSong, I. -W., Vo, H. H., Chen, Y. -S., Baysal, M. A., Kahle, M., Johnson, A., & Tsimberidou, A. M. (2023). Precision Oncology: Evolving Clinical Trials across Tumor Types. Cancers, 15(7), 1967. https://doi.org/10.3390/cancers15071967