


Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. Whole-Genome Sequencing and DNA Methylation Profiling
2.3. Droplet Digital PCR
- (i)
- events typical for MB according to the WHO Classification of Central Nervous System Tumors, 5th edition [5];
- (ii)
- (iii)
- novel, tumor-specific junction sequences resulting from SVs.
3. Results
3.1. Participants’ Characteristics
3.2. Target Identification in WGS Tumor Data
3.3. ddPCR Assay Performance; Sensitivity and False-Positive Rates
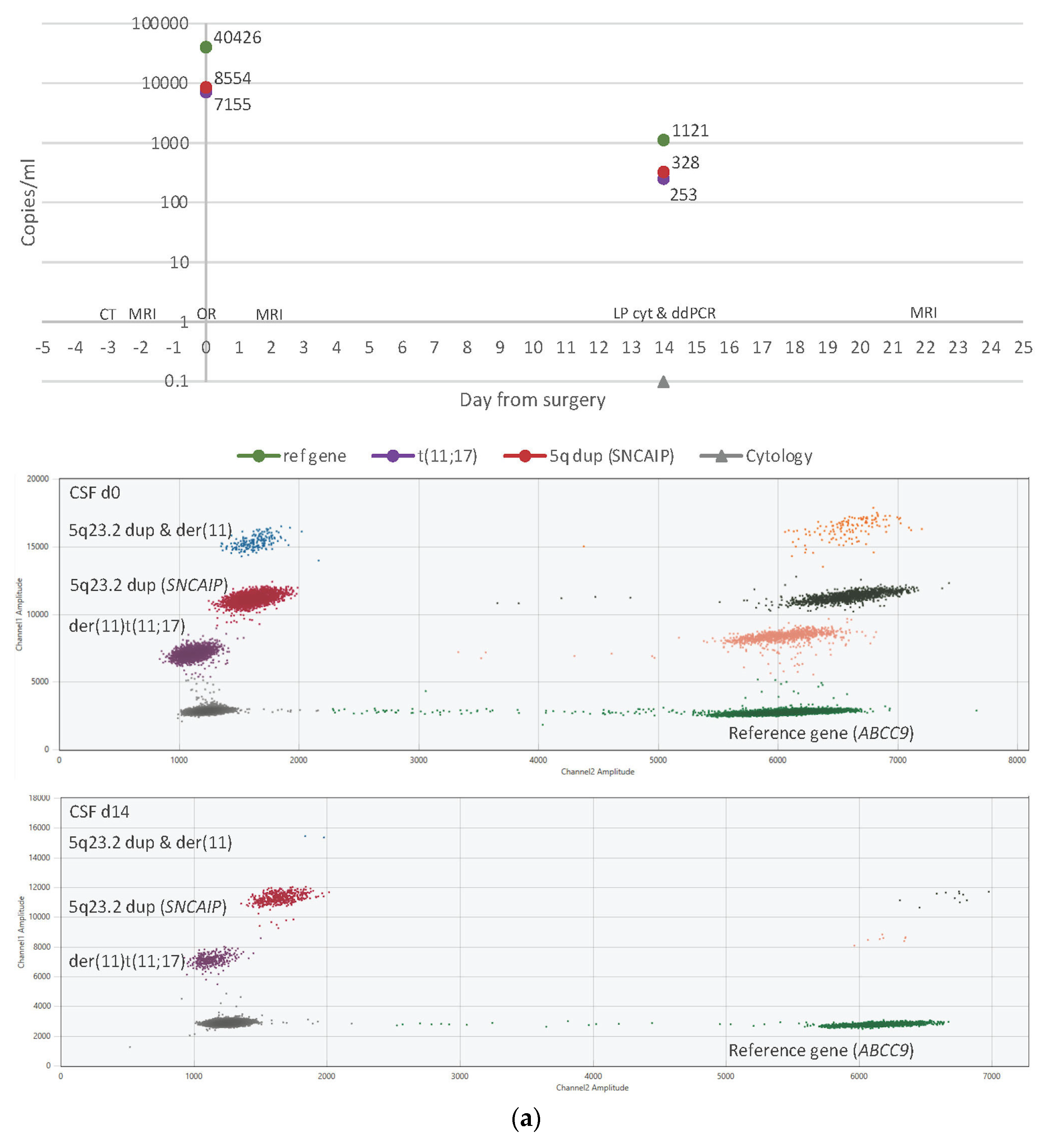
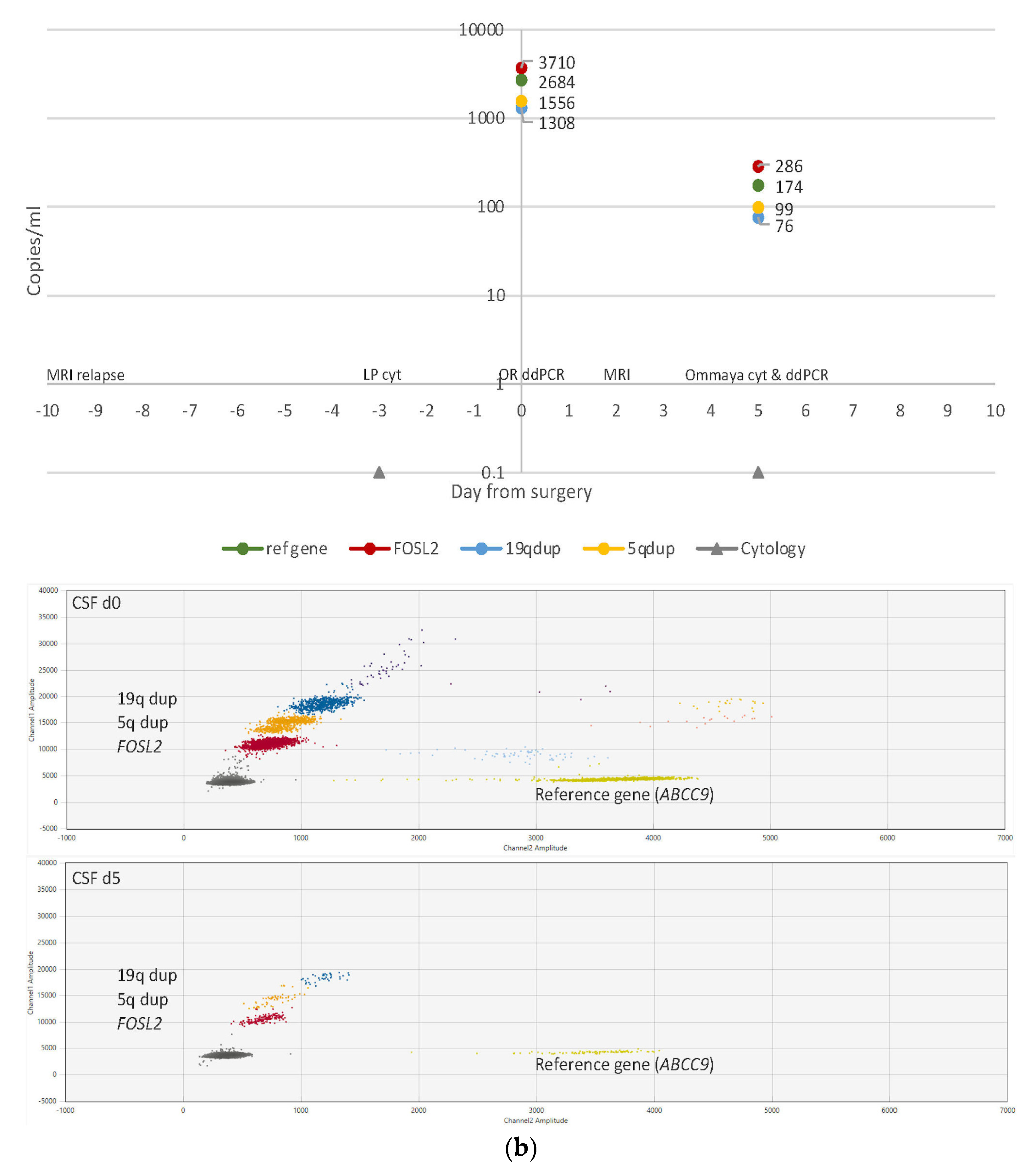
3.4. ctDNA Detection in CSF Samples
3.5. ctDNA Detection in Plasma Samples
3.6. CSF Cytology
3.7. Imaging Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Ryan, K.; Edelson, J.; Neff, C.; Cioffi, G.; A Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Pediatric Brain Tumor Foundation Childhood and Adolescent Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2022, 24, iii1–iii38. [Google Scholar] [CrossRef] [PubMed]
- Khanna, V.; Achey, R.L.; Ostrom, Q.; Block-Beach, H.; Kruchko, C.; Barnholtz-Sloan, J.S.; de Blank, P. Incidence and survival trends for medulloblastomas in the United States from 2001 to 2013. J. Neuro-Oncol. 2017, 135, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6. [Google Scholar]
- Mynarek, M.; Milde, T.; Padovani, L.; Janssens, G.O.; Kwiecien, R.; Mosseri, V.; Clifford, S.C.; Doz, F.; Rutkowski, S. SIOP PNET5 MB Trial: History and Concept of a Molecularly Stratified Clinical Trial of Risk-Adapted Therapies for Standard-Risk Medulloblastoma. Cancers 2021, 13, 6077. [Google Scholar] [CrossRef]
- HIT-MED Guidance for Patients with Newly Diagnosed Medulloblastoma Ependymoma and Pineoblastoma Version 5.1. HIT-MED Study Centre at the University Medical Center Hamburg-Eppendorf, Germany. Available online: https://www.skion.nl/workspace/uploads/HIT-MED-Guidance-Final-Version-5-1.pdf (accessed on 9 March 2023).
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Gajjar, A.; Robinson, G.W.; Smith, K.S.; Lin, T.; Merchant, T.E.; Chintagumpala, M.; Mahajan, A.; Su, J.; Bouffet, E.; Bartels, U.; et al. Outcomes by Clinical and Molecular Features in Children with Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J. Clin. Oncol. 2021, 39, 822–835. [Google Scholar] [CrossRef]
- Kortmann, R.-D.; Kühl, J.; Timmermann, B.; Mittler, U.; Urban, C.; Budach, V.; Richter, E.; Willich, N.; Flentje, M.; Berthold, F.; et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: Results of the german prospective randomized trial hit ’91. Int. J. Radiat. Oncol. 2000, 46, 269–279. [Google Scholar] [CrossRef]
- Lannering, B.; Rutkowski, S.; Doz, F.; Pizer, B.; Gustafsson, G.; Navajas, A.; Massimino, M.; Reddingius, R.; Benesch, M.; Carrie, C.; et al. Hyperfractionated Versus Conventional Radiotherapy Followed by Chemotherapy in Standard-Risk Medulloblastoma: Results From the Randomized Multicenter HIT-SIOP PNET 4 Trial. J. Clin. Oncol. 2012, 30, 3187–3193. [Google Scholar] [CrossRef]
- Ribi, K.; Relly, C.; Landolt, M.A.; Alber, F.D.; Boltshauser, E.; Grotzer, M.A. Outcome of Medulloblastoma in Children: Long-Term Complications and Quality of Life. Neuropediatrics 2005, 36, 357–365. [Google Scholar] [CrossRef]
- Oyefiade, A.; Paltin, I.; De Luca, C.R.; Hardy, K.K.; Grosshans, D.R.; Chintagumpala, M.; Mabbott, D.J.; Kahalley, L.S. Cognitive Risk in Survivors of Pediatric Brain Tumors. J. Clin. Oncol. 2021, 39, 1718–1726. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Rosenquist, R.; Cuppen, E.; Buettner, R.; Caldas, C.; Dreau, H.; Elemento, O.; Frederix, G.; Grimmond, S.; Haferlach, T.; Jobanputra, V.; et al. Clinical utility of whole-genome sequencing in precision oncology. Semin. Cancer Biol. 2022, 84, 32–39. [Google Scholar] [CrossRef]
- Cuppen, E.; Elemento, O.; Rosenquist, R.; Nikic, S.; Ijzerman, M.; Zaleski, I.D.; Frederix, G.; Levin, L.; Mullighan, C.G.; Buettner, R.; et al. Implementation of Whole-Genome and Transcriptome Sequencing Into Clinical Cancer Care. JCO Precis. Oncol. 2022. [Google Scholar] [CrossRef]
- Fouladi, M.; Gajjar, A.; Boyett, J.M.; Walter, A.W.; Thompson, S.J.; Merchant, T.E.; Jenkins, J.J.; Langston, J.W.; Liu, A.; Kun, L.E.; et al. Comparison of CSF Cytology and Spinal Magnetic Resonance Imaging in the Detection of Leptomeningeal Disease in Pediatric Medulloblastoma or Primitive Neuroectodermal Tumor. J. Clin. Oncol. 1999, 17, 3234–3237. [Google Scholar] [CrossRef]
- Terterov, S.; Krieger, M.D.; Bowen, I.; McComb, J.G. Evaluation of intracranial cerebrospinal fluid cytology in staging pediatric medulloblastomas, supratentorial primitive neuroectodermal tumors, and ependymomas. J. Neurosurg. Pediatr. 2010, 6, 131–136. [Google Scholar] [CrossRef] [Green Version]
- ALLTogether Study—A Treatment Protocol for Participants 1–45 Years with Acute Lymphoblastic Leukaemia. Available online: https://clinicaltrials.gov/ct2/show/NCT03911128 (accessed on 16 January 2023).
- Oncology NSoPHa. NOPHO-DBH AML 2012 Protocol v2.1. 2013:68. EUdract Number 2012-002934-35. Available online: https://clinicaltrials.gov/ct2/show/NCT00819351 (accessed on 16 January 2023).
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minimal Residual Disease–Directed Adjuvant Therapy for Patients with Early-Stage Colon Cancer: CIRCULATE-US. Oncology 2022, 36, 604–608. [CrossRef]
- Pellini, B.; Chaudhuri, A.A. Circulating Tumor DNA Minimal Residual Disease Detection of Non–Small-Cell Lung Cancer Treated With Curative Intent. J. Clin. Oncol. 2022, 40, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; A Memoli, V.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Biomark. Prev. 1994, 3, 67–71. [Google Scholar]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol. Commun. 2017, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- E Piccioni, D.; Achrol, A.S.; A Kiedrowski, L.; Banks, K.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- García-Romero, N.; Carrión-Navarro, J.; Areal-Hidalgo, P.; de Mendivil, A.O.; Asensi-Puig, A.; Madurga, R.; Núñez-Torres, R.; González-Neira, A.; Belda-Iniesta, C.; González-Rumayor, V.; et al. BRAF V600E Detection in Liquid Biopsies from Pediatric Central Nervous System Tumors. Cancers 2019, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, S.; Lee, M.; Yin, Y.; Li, J.; Zhou, Y.; Ballester, L.Y.; Esquenazi, Y.; Dashwood, R.H.; Davies, P.J.A.; et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 2020, 6, eabb5427. [Google Scholar] [CrossRef]
- Escudero, L.; Llort, A.; Arias, A.; Diaz-Navarro, A.; Martínez-Ricarte, F.; Rubio-Perez, C.; Mayor, R.; Caratù, G.; Martínez-Sáez, E.; Vázquez-Méndez, É.; et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 2020, 11, 5736. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Ren, S.; Liu, Y.; Zhang, J.; Li, S.; Gao, W.; Gong, X.; Liu, J.; Wang, Y.; et al. Exploring genetic alterations in circulating tumor DNA from cerebrospinal fluid of pediatric medulloblastoma. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Pagès, M.; Rotem, D.; Gydush, G.; Reed, S.; Rhoades, J.; Ha, G.; Lo, C.; Fleharty, M.; Duran, M.; Jones, R.; et al. Liquid biopsy detection of genomic alterations in pediatric brain tumors from cell-free DNA in peripheral blood, CSF, and urine. Neuro-Oncology 2022, 24, 1352–1363. [Google Scholar] [CrossRef]
- Miller, A.M.; Szalontay, L.; Bouvier, N.; Hill, K.; Ahmad, H.; Rafailov, J.; Lee, A.J.; Rodriguez-Sanchez, M.I.; Yildirim, O.; Patel, A.; et al. Next-generation sequencing of cerebrospinal fluid for clinical molecular diagnostics in pediatric, adolescent and young adult brain tumor patients. Neuro-Oncology 2022, 24, 1763–1772. [Google Scholar] [CrossRef]
- Liu, A.P.; Smith, K.S.; Kumar, R.; Paul, L.; Bihannic, L.; Lin, T.; Maass, K.K.; Pajtler, K.W.; Chintagumpala, M.; Su, J.M.; et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021, 39, 1519–1530.e4. [Google Scholar] [CrossRef]
- Koss, L.G. Diagnostic Cytology and Its Histopathologic Bases, 2nd ed.; J. B. Lippincott & Co.: Philadelphia, PA, USA, 1968. [Google Scholar]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; De Ståhl, T.D.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Research 2020, 9, 63. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. Available online: https://grch37.ensembl.org/info/docs/tools/vep/index.html (accessed on 22 March 2022). [CrossRef] [PubMed] [Green Version]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Wet-Lab Validated ddPCR Assays for Mutation Detection and Copy Number Determination. Bio-Rad Laboratories, Inc. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_7144.pdf (accessed on 8 April 2022).
- Droplet Digital PCR ASSAY DESIGN ENGINE, Mutation Detection. Bio-Rad Laboratories, Inc. Available online: https://www.bio-rad.com/digital-assays/assays-create/mutation (accessed on 13 April 2022).
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [Green Version]
- Rare Mutation Detection Best Practices Guidelines Droplet Digital™ PCR. Bio-Rad Laboratories, Inc. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6628.pdf (accessed on 22 March 2022).
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Droplet Digital™ PCR Applications Guide. Bio-Rad Laboratories, Inc. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf (accessed on 22 March 2022).
- Blattner-Johnson, M.; Jones, D.T.; Pfaff, E. Precision medicine in pediatric solid cancers. Semin. Cancer Biol. 2022, 84, 214–227. [Google Scholar] [CrossRef]
- Fioretos, T.; Wirta, V.; Cavelier, L.; Berglund, E.; Friedman, M.; Akhras, M.; Botling, J.; Ehrencrona, H.; Engstrand, L.; Helenius, G.; et al. Implementing precision medicine in a regionally organized healthcare system in Sweden. Nat. Med. 2022, 28, 1980–1982. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Vuong, T.A.; Jeong, H.-J.; Lee, H.-J.; Kim, B.-G.; Leem, Y.-E.; Cho, H.; Kang, J.-S. PRMT7 methylates and suppresses GLI2 binding to SUFU thereby promoting its activation. Cell Death Differ. 2019, 27, 15–28. [Google Scholar] [CrossRef]
- HGNC Database. HUGO Gene Nomenclature Committee (HGNC), European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom. Available online: www.genenames.org (accessed on 1 April 2022).
- Gao, L.; Guo, Y.; Zeng, J.; Ma, F.; Luo, J.; Zhu, H.; Xia, S.; Wei, K.; Chen, G. The expression, significance and function of cancer susceptibility candidate 9 in lung squamous cell carcinoma: A bioinformatics and in vitro investigation. Int. J. Oncol. 2019, 54, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, D.; Wang, Y.; Ren, F.; Pang, S.; Wang, D.; Xu, S. FOSL2 Positively Regulates TGF-β1 Signalling in Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e112150. [Google Scholar] [CrossRef]
- Nakayama, T.; Hieshima, K.; Arao, T.; Jin, Z.; Nagakubo, D.; Shirakawa, A.-K.; Yamada, Y.; Fujii, M.; Oiso, N.; Kawada, A.; et al. Aberrant expression of Fra-2 promotes CCR4 expression and cell proliferation in adult T-cell leukemia. Oncogene 2007, 27, 3221–3232. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhang, Z.; Liu, C.; Jin, C.; Zhang, J.; Miao, X.; Jia, L. B4GALT1 gene knockdown inhibits the hedgehog pathway and reverses multidrug resistance in the human leukemia K562/adriamycin-resistant cell line. IUBMB Life 2012, 64, 889–900. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, H.; Wei, W.; Ji, D.; Song, X.; Sun, J.; Zhang, J.; Jia, L. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis. 2013, 4, e654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Han, L.; Chen, Y.; He, F.; Sun, B.; Kamar, S.; Zhang, Y.; Yang, Y.; Wang, C.; Yang, Z. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.W.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2012, 125, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.W.; Northcott, P.A.; Kool, M.; Pfister, S.M. The Role of Chromatin Remodeling in Medulloblastoma. Brain Pathol. 2013, 23, 193–199. [Google Scholar] [CrossRef]
- Qin, Z.; Ren, F.; Xu, X.; Ren, Y.; Li, H.; Wang, Y.; Zhai, Y.; Chang, Z. ZNF536, a Novel Zinc Finger Protein Specifically Expressed in the Brain, Negatively Regulates Neuron Differentiation by Repressing Retinoic Acid-Induced Gene Transcription. Mol. Cell. Biol. 2009, 29, 3633–3643. [Google Scholar] [CrossRef] [Green Version]
- Natrajan, R.; Mackay, A.; Wilkerson, P.M.; Lambros, M.B.; Wetterskog, D.; Arnedos, M.; Shiu, K.-K.; Geyer, F.C.; Langerød, A.; Kreike, B.; et al. Functional characterization of the 19q12 amplicon in grade III breast cancers. Breast Cancer Res. 2012, 14, R53. [Google Scholar] [CrossRef] [Green Version]
- Bickert, A.; Ginkel, C.; Kol, M.; Vom Dorp, K.; Jastrow, H.; Degen, J.; Jacobs, R.L.; Vance, D.E.; Winterhager, E.; Jiang, X.-C.; et al. Functional characterization of enzymes catalyzing ceramide phosphoethanolamine biosynthesis in mice. J. Lipid Res. 2015, 56, 821–835. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000156671-SAMD8 (accessed on 9 January 2023).
- Tafesse, F.G.; Vacaru, A.M.; Bosma, E.F.; Hermansson, M.; Jain, A.; Hilderink, A.; Somerharju, P.; Holthuis, J. Sphingomyelin synthase-related protein SMSr is a suppressor of ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2013, 127, 445–454. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| UPN | Demographics | Clinical Features | Molecular Features | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (Y) | Gender | Tumor Location | Size (cm) | Histology | Treatment Protocol | Mutations Somatic | Germline | CNAs | Subgroup | Methylation Subtype | |
| 1 | 14 | M | 4th ventricle | 4 | C | PNET5 | — | — | ¾ | VIII |
| 9 | 10 | F | 4th ventricle | 3.5 × 3 × 4 | C | PNET5 | CTNNB1 | — | 6- | WNT |
| 13 | 7 | F | 4th ventricle | 4.5 × 4.5 × 4 | C | PNET5 | CTNNB1 | — | 6- | WNT |
| 15p | 5 | M | 4th ventricle | 4.5 × 4 × 4 | C | PNET5 | — | — | ¾ | V |
| 15 r/m | 6 | M | 4th ventricle, base of scull, spinal cord | NA/0.4–0.6/0.7 | C | MEMMAT | — | — | ¾ | III |
| 19p | 4 | F | LCH | 6 × 5 × 3.8 | C | PNET5 | — | — | SHH | A |
| 19r | 5 | F | LCH | 0.4/0.6/0.2 | C | TOTEM | — | — | SHH | A |
| 21 | 4 | M | 4th ventricle | 4.5 × 4 × 3.7 | C | HIT Medguidance | — | — | ¾ | VI |
| 22 | 5 | M | 4th ventricle | 4.5 × 5 × 4 | C | PNET5 | — | — | ¾ | VI * |
| 24 | 1 | F | RCH | 2.5 | D/N | HIT-SKK | — | — | SHH | B |
| 27 | 1 | M | Vermis | 4.5 × 3.5 × 4 | D/N | HIT-SKK | — | SUFU | — | SHH | B |
| 31 | 2 | F | 4th ventricle | 3.5 × 3 × 3.5 | N | HIT Medguidance | SMO, KMT2D | — | — | SHH | B |
| 35 | 5 | M | 4th ventricle | 2.4 | C | PNET5 | CTNNB1 | — | — | WNT |
| 43 | 15 | F | 4th ventricle | 4 × 3.5 × 2.5 | C | NA | KDM6A | — | — | ¾ | NA |
| UPN | Target | Target Position GRCh38/hg38 | Amplicon Size | LoD | FPR |
|---|---|---|---|---|---|
| (bp) | (pg) | Events/Well | |||
| 1 | der(11)t(11;17)(p11.2;q23.1) | chr11:46,349,051 | 98 | 100 | 0 |
| 5q23.2 tandem duplication (SNCAIP) | chr5:122,518,910 | 100 | 100 | 0 | |
| 9 | CTNNB1 c.100G>A, p.Gly34Arg * | chr3:41,224,612 | 62 | 100 | 0.17 |
| CSNK2B c.419G>T, p.Cys140Phe | chr6:31,669,370 | 64 | 100 | 0 | |
| 1p21.1 intergenic tandem duplication | chr1:105,150,201 | 89 | 100 | 0 | |
| 13 | CTNNB1 c.101G>A, p.Gly34Glu * | chr3:41,224,613 | 64 | 100 | 0.58 |
| PIK3CA c.3140A>G, p.His1047Arg | chr3:179,234,297 | 80 | 100 | 0 | |
| 15 | FOSL2 c.383G>A, p.Arg128His | chr2:28,408,787 | 62 | 10 | 0 |
| 19q12 tandem duplication (ZNF536) | chr19:30,403,430 | 116 | 100 | 0 | |
| 5q13.3 tandem duplication (HEXB) | chr5:74,152,786 | 91 | 100 | 0 | |
| 19 | MAX c.179G>A, p.Arg60Gln | chr14:65,078,029 | 69 | 100 | 0.25 |
| PTCH1 c.2287dup, p.Val763GlyfsTer27 | chr9:95,467,388 | 63 | 100 | 0.25 | |
| 21 | B4GALT1 c.421_428dup p.Glu144CysfsTer2 | chr9:33,135,408 | 64 | 10 | 0 |
| 10q22.2 amp (SAMD8) | chr10:73,926,074 | 87 | 10 | 0 | |
| 22 | KDM5D c.1642G>C, p.Asp548His | chrY:19,720,946 | 64 | 100 | 0 |
| 7q21.2 amp (CDK6) | chr7:92,501,482 | 92 | 10 | 0 | |
| 24 | PRMT7 c.224C>T, p.Thr75Met | chr16:68,324,774 | 62 | 10 | 0.5 |
| 27 | KMT2D c.13825C>T, p.Gln4609Ter | chr12:49,030,615 | 98 | 100 | 0.6 |
| OFD1 c.1411+1G>A | chrX:13,756,768 | 65 | 10 | 0.25 | |
| 31 | KMT2D c.7933C>T, p.Arg2645Ter | chr12:49,039,837 | 68 | 100 | 0.75 |
| SMO c.1247_1248delinsAA, p.Gly416Glu * | chr7:129,206,570 | 61 | 100 | 1.58 | |
| 35 | CTNNB1, c.101G>A, p.Gly34Glu * | chr3:41,224,613 | 64 | 100 | 0.17 |
| 13q14.2 tandem duplication (CAB39L) | chr13:49,304,384 | 112 | 100 | 0 | |
| 43 | KDM6A c.4129C>T, p.Gln1377Ter * | chrX:45,110,202 | 68 | 100 | 0.08 |
| Xp22.11 tandem duplication (KLHL15) | chrX:24,005,865 | 90 | 100 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arthur, C.; Jylhä, C.; de Ståhl, T.D.; Shamikh, A.; Sandgren, J.; Rosenquist, R.; Nordenskjöld, M.; Harila, A.; Barbany, G.; Sandvik, U.; et al. Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma. Cancers 2023, 15, 1972. https://doi.org/10.3390/cancers15071972
Arthur C, Jylhä C, de Ståhl TD, Shamikh A, Sandgren J, Rosenquist R, Nordenskjöld M, Harila A, Barbany G, Sandvik U, et al. Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma. Cancers. 2023; 15(7):1972. https://doi.org/10.3390/cancers15071972
Chicago/Turabian StyleArthur, Cecilia, Cecilia Jylhä, Teresita Díaz de Ståhl, Alia Shamikh, Johanna Sandgren, Richard Rosenquist, Magnus Nordenskjöld, Arja Harila, Gisela Barbany, Ulrika Sandvik, and et al. 2023. "Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma" Cancers 15, no. 7: 1972. https://doi.org/10.3390/cancers15071972
APA StyleArthur, C., Jylhä, C., de Ståhl, T. D., Shamikh, A., Sandgren, J., Rosenquist, R., Nordenskjöld, M., Harila, A., Barbany, G., Sandvik, U., & Tham, E. (2023). Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma. Cancers, 15(7), 1972. https://doi.org/10.3390/cancers15071972