

The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
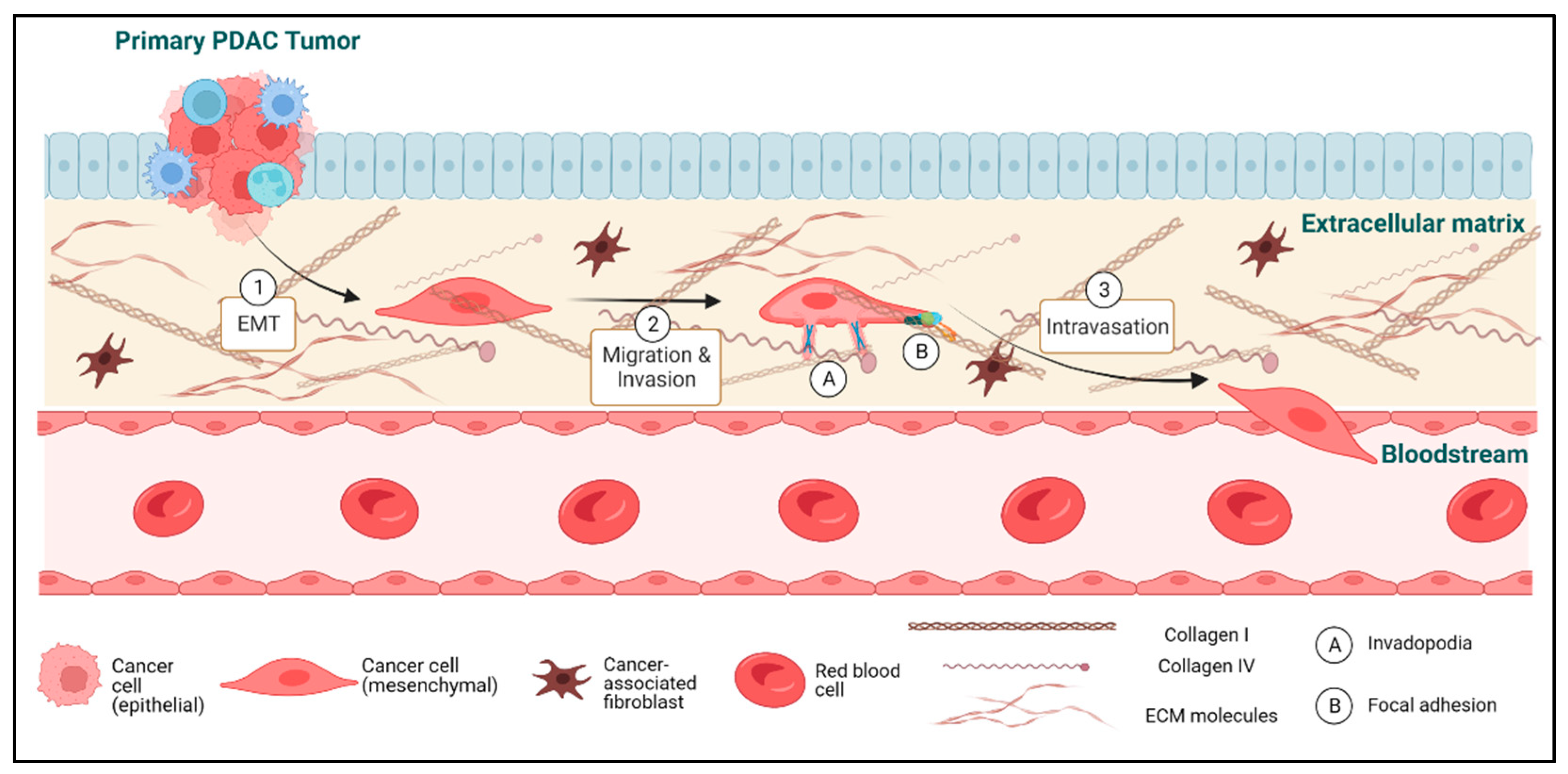
2. Epithelial-to-Mesenchymal Transition in PDAC
2.1. The Complex Role of EMT in Metastasis
2.2. Novel Mechanistic Insights on EMT in PDAC: E-Cadherin
2.3. Role of the TME in EMT
3. Invadopodia and Protease-Mediated Degradation of the ECM
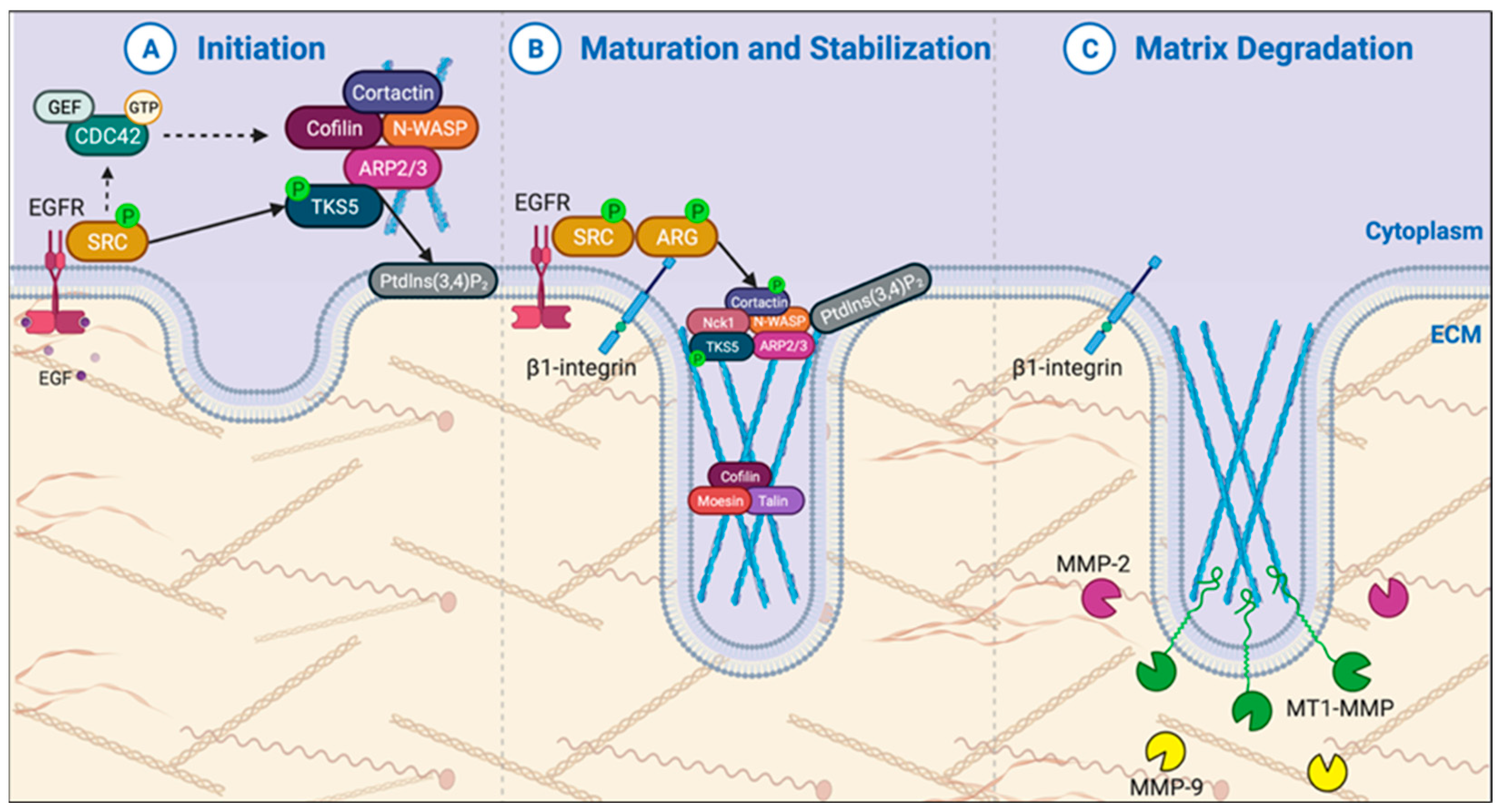
3.1. Novel Mechanistic Insights on Invadopodia in PDAC
3.1.1. Invadopodia Initiation & Formation
3.1.2. Invadopodia Maturation & Stabilization
3.1.3. Invadopodia Proteinase Activity
3.2. Role of the TME in Invadopodial ECM Degradation
3.3. Invadopodia and Matrix Degradation: Final Thoughts
4. Tumor Cell Migration via Lamellipodia and Focal Adhesions
4.1. Lamellipodia and Filopodia in PDAC Cells
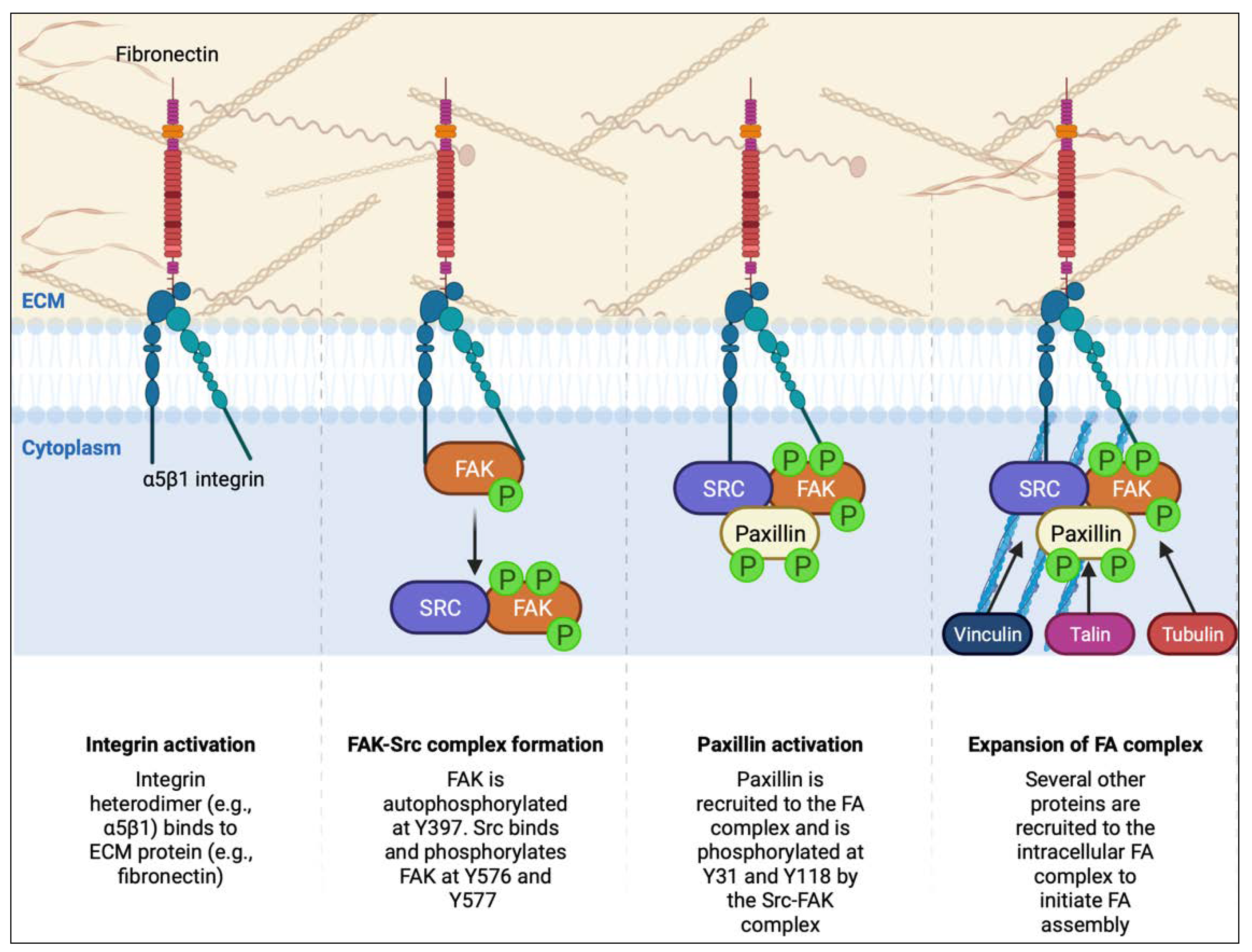
4.2. Focal Adhesion Dynamics in PDAC
4.2.1. Integrin Trafficking
4.2.2. FAK and Paxillin Regulation
4.2.3. Microtubule Dynamics in Focal Adhesion Turnover
4.3. Role of the TME in PDAC Focal Adhesions
4.4. Focal Adhesions: Final Thoughts
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.P.t.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Sinha, S.; Leach, S.D. New insights in the development of pancreatic cancer. Curr. Opin. Gastroenterol. 2016, 32, 394–400. [Google Scholar] [CrossRef]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Niederhuber, J.E.; Brennan, M.F.; Menck, H.R. The National Cancer Data Base report on pancreatic cancer. Cancer 1995, 76, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummers, W.S.; Groen, J.V.; Sibinga Mulder, B.G.; Farina-Sarasqueta, A.; Morreau, J.; Putter, H.; van de Velde, C.J.; Vahrmeijer, A.L.; Bonsing, B.A.; Mieog, J.S.; et al. Impact of resection margin status on recurrence and survival in pancreatic cancer surgery. Br. J. Surg. 2019, 106, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massague, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Wells, A.; Griffith, L.; Wells, J.Z.; Taylor, D.P. The dormancy dilemma: Quiescence versus balanced proliferation. Cancer Res. 2013, 73, 3811–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef]
- Sundahl, N.; Duprez, F.; Ost, P.; De Neve, W.; Mareel, M. Effects of radiation on the metastatic process. Mol. Med. 2018, 24, 16. [Google Scholar] [CrossRef] [Green Version]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Li, W.; Kang, Y. Probing the Fifty Shades of EMT in Metastasis. Trends Cancer 2016, 2, 65–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Shamir, E.R.; Pappalardo, E.; Jorgens, D.M.; Coutinho, K.; Tsai, W.T.; Aziz, K.; Auer, M.; Tran, P.T.; Bader, J.S.; Ewald, A.J. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J. Cell Biol. 2014, 204, 839–856. [Google Scholar] [CrossRef]
- Palamaris, K.; Felekouras, E.; Sakellariou, S. Epithelial to Mesenchymal Transition: Key Regulator of Pancreatic Ductal Adenocarcinoma Progression and Chemoresistance. Cancers 2021, 13, 5532. [Google Scholar] [CrossRef]
- Paul, M.C.; Schneeweis, C.; Falcomata, C.; Shan, C.; Rossmeisl, D.; Koutsouli, S.; Klement, C.; Zukowska, M.; Widholz, S.A.; Jesinghaus, M.; et al. Non-canonical functions of SNAIL drive context-specific cancer progression. Nat. Commun. 2023, 14, 1201. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Sampson, V.B.; David, J.M.; Puig, I.; Patil, P.U.; de Herreros, A.G.; Thomas, G.V.; Rajasekaran, A.K. Wilms’ tumor protein induces an epithelial-mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS ONE 2014, 9, e102041. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Carstens, J.L.; Yang, S.; Correa de Sampaio, P.; Zheng, X.; Barua, S.; McAndrews, K.M.; Rao, A.; Burks, J.K.; Rhim, A.D.; Kalluri, R. Stabilized epithelial phenotype of cancer cells in primary tumors leads to increased colonization of liver metastasis in pancreatic cancer. Cell Rep. 2021, 35, 108990. [Google Scholar] [CrossRef]
- Beerling, E.; Oosterom, I.; Voest, E.; Lolkema, M.; van Rheenen, J. Intravital characterization of tumor cell migration in pancreatic cancer. Intravital 2016, 5, e1261773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Molnar, B.; Ladanyi, A.; Tanko, L.; Sreter, L.; Tulassay, Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin. Cancer Res. 2001, 7, 4080–4085. [Google Scholar]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2017, 11, 5–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Tanaka, S.; Akiyama, Y.; Shimada, S.; Adikrisna, R.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Dominant Expression of DCLK1 in Human Pancreatic Cancer Stem Cells Accelerates Tumor Invasion and Metastasis. PLoS ONE 2016, 11, e0146564. [Google Scholar] [CrossRef] [Green Version]
- Pinner, S.; Sahai, E. Imaging amoeboid cancer cell motility in vivo. J. Microsc. 2008, 231, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Graziani, V.; Rodriguez-Hernandez, I.; Maiques, O.; Sanz-Moreno, V. The amoeboid state as part of the epithelial-to-mesenchymal transition programme. Trends Cell Biol. 2022, 32, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Jang, S.D.; Kim, H.; Chung, S.; Park, J.K.; Kuh, H.J. Phenotypic Heterogeneity and Plasticity of Cancer Cell Migration in a Pancreatic Tumor Three-Dimensional Culture Model. Cancers 2020, 12, 1305. [Google Scholar] [CrossRef]
- Facoetti, A.; Di Gioia, C.; Pasi, F.; Di Liberto, R.; Corbella, F.; Nano, R.; Ciocca, M.; Valvo, F.; Orecchia, R. Morphological Analysis of Amoeboid-Mesenchymal Transition Plasticity After Low and High LET Radiation on Migrating and Invading Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 4585–4591. [Google Scholar] [CrossRef]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Bruser, L.; Bogdan, S. Adherens Junctions on the Move-Membrane Trafficking of E-Cadherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a029140. [Google Scholar] [CrossRef] [Green Version]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Hapach, L.A.; Carey, S.P.; Schwager, S.C.; Taufalele, P.V.; Wang, W.; Mosier, J.A.; Ortiz-Otero, N.; McArdle, T.J.; Goldblatt, Z.E.; Lampi, M.C.; et al. Phenotypic Heterogeneity and Metastasis of Breast Cancer Cells. Cancer Res. 2021, 81, 3649–3663. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Buck, C.A.; Bechtol, K.B.; Damsky, C.H. Soluble 80-kd fragment of cell-CAM 120/80 disrupts cell-cell adhesion. J. Cell. Biochem. 1987, 34, 187–202. [Google Scholar] [CrossRef]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. 2012, 17, 1948–1964. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.K.; Ramani, V.C.; Hennings, L.; Haun, R.S. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 2007, 109, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Perusina Lanfranca, M.; Zhang, Y.; Girgis, A.; Kasselman, S.; Lazarus, J.; Kryczek, I.; Delrosario, L.; Rhim, A.; Koneva, L.; Sartor, M.; et al. Interleukin 22 Signaling Regulates Acinar Cell Plasticity to Promote Pancreatic Tumor Development in Mice. Gastroenterology 2020, 158, 1417–1432.e11. [Google Scholar] [CrossRef]
- Tang, D.; Zhang, J.; Yuan, Z.; Zhang, H.; Chong, Y.; Huang, Y.; Wang, J.; Xiong, Q.; Wang, S.; Wu, Q.; et al. PSC-derived Galectin-1 inducing epithelial-mesenchymal transition of pancreatic ductal adenocarcinoma cells by activating the NF-kappaB pathway. Oncotarget 2017, 8, 86488–86502. [Google Scholar] [CrossRef] [Green Version]
- Tekin, C.; Aberson, H.L.; Waasdorp, C.; Hooijer, G.K.J.; de Boer, O.J.; Dijk, F.; Bijlsma, M.F.; Spek, C.A. Macrophage-secreted MMP9 induces mesenchymal transition in pancreatic cancer cells via PAR1 activation. Cell. Oncol. 2020, 43, 1161–1174. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Liao, Q.; Zhao, J.; Hu, Y.; You, L.; Lu, Z.; Jia, C.; Wei, Y.; Zhao, Y. High expression of interleukin-22 and its receptor predicts poor prognosis in pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Gimona, M.; Buccione, R.; Courtneidge, S.A.; Linder, S. Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 2008, 20, 235–241. [Google Scholar] [CrossRef]
- Beaty, B.T.; Condeelis, J. Digging a little deeper: The stages of invadopodium formation and maturation. Eur. J. Cell Biol. 2014, 93, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Baik, M.; Byers, J.T.; Chen, K.T.; French, S.W.; Diaz, B. TKS5-positive invadopodia-like structures in human tumor surgical specimens. Exp. Mol. Pathol. 2019, 106, 17–26. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [Green Version]
- Botta, G.P.; Reginato, M.J.; Reichert, M.; Rustgi, A.K.; Lelkes, P.I. Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1. Mol. Cancer Res. 2012, 10, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Hu, J.; Liu, Y.; Li, L.; Li, Y.; Sun, B.; Kong, R. Invadopodia: A potential target for pancreatic cancer therapy. Crit. Rev. Oncol. Hematol. 2021, 159, 103236. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H. Pathological roles of invadopodia in cancer invasion and metastasis. Eur. J. Cell Biol. 2012, 91, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Cervero, P.; Eddy, R.; Condeelis, J. Mechanisms and roles of podosomes and invadopodia. Nat. Rev. Mol. Cell Biol. 2022, 24, 86–106. [Google Scholar] [CrossRef]
- Sharma, V.P.; Eddy, R.; Entenberg, D.; Kai, M.; Gertler, F.B.; Condeelis, J. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr. Biol. 2013, 23, 2079–2089. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Lorenz, M.; Kempiak, S.; Sarmiento, C.; Coniglio, S.; Symons, M.; Segall, J.; Eddy, R.; Miki, H.; Takenawa, T.; et al. Molecular mechanisms of invadopodium formation: The role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 2005, 168, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neel, N.F.; Rossman, K.L.; Martin, T.D.; Hayes, T.K.; Yeh, J.J.; Der, C.J. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol. Cell Biol. 2012, 32, 1374–1386. [Google Scholar] [CrossRef] [Green Version]
- Razidlo, G.L.; Schroeder, B.; Chen, J.; Billadeau, D.D.; McNiven, M.A. Vav1 as a central regulator of invadopodia assembly. Curr. Biol. 2014, 24, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Stock, K.; Borrink, R.; Mikesch, J.H.; Hansmeier, A.; Rehkamper, J.; Trautmann, M.; Wardelmann, E.; Hartmann, W.; Sperveslage, J.; Steinestel, K. Overexpression and Tyr421-phosphorylation of cortactin is induced by three-dimensional spheroid culturing and contributes to migration and invasion of pancreatic ductal adenocarcinoma (PDAC) cells. Cancer Cell Int. 2019, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Lian, E.Y.; Hyndman, B.D.; Moodley, S.; Maritan, S.M.; Mulligan, L.M. RET isoforms contribute differentially to invasive processes in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 6493–6510. [Google Scholar] [CrossRef]
- Oser, M.; Yamaguchi, H.; Mader, C.C.; Bravo-Cordero, J.J.; Arias, M.; Chen, X.; Desmarais, V.; van Rheenen, J.; Koleske, A.J.; Condeelis, J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 2009, 186, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Gligorijevic, B.; Wyckoff, J.; Yamaguchi, H.; Wang, Y.; Roussos, E.T.; Condeelis, J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 2012, 125, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-Sastre, A.; Desztics, J.; Dantes, Z.; Schulte, K.; Ensarioglu, H.K.; Bassey-Archibong, B.; Ollinger, R.; Engleiter, T.; Rayner, L.; Einwachter, H.; et al. Loss of Wasl improves pancreatic cancer outcome. JCI Insight 2020, 5, 127275. [Google Scholar] [CrossRef]
- Guo, J.C.; Li, J.; Zhao, Y.P.; Zhou, L.; Cui, Q.C.; Zhou, W.X.; Zhang, T.P.; You, L.; Shu, H. N-wasp in pancreatic ductal adenocarcinoma: Associations with perineural invasion and poor prognosis. World J. Surg. 2014, 38, 2126–2131. [Google Scholar] [CrossRef]
- Juin, A.; Spence, H.J.; Martin, K.J.; McGhee, E.; Neilson, M.; Cutiongco, M.F.A.; Gadegaard, N.; Mackay, G.; Fort, L.; Lilla, S.; et al. N-WASP Control of LPAR1 Trafficking Establishes Response to Self-Generated LPA Gradients to Promote Pancreatic Cancer Cell Metastasis. Dev. Cell 2019, 51, 431–445.e7. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, H.E.; Teppo, S.; Niemela, S.; Kallioniemi, A. Silencing of the ARP2/3 complex disturbs pancreatic cancer cell migration. Anticancer Res. 2013, 33, 45–52. [Google Scholar]
- Li, A.; Dawson, J.C.; Forero-Vargas, M.; Spence, H.J.; Yu, X.; Konig, I.; Anderson, K.; Machesky, L.M. The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr. Biol. 2010, 20, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Morton, J.P.; Ma, Y.; Karim, S.A.; Zhou, Y.; Faller, W.J.; Woodham, E.F.; Morris, H.T.; Stevenson, R.P.; Juin, A.; et al. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology 2014, 146, 1386–1396.e17. [Google Scholar] [CrossRef] [Green Version]
- Welsch, T.; Keleg, S.; Bergmann, F.; Bauer, S.; Hinz, U.; Schmidt, J. Actinin-4 expression in primary and metastasized pancreatic ductal adenocarcinoma. Pancreas 2009, 38, 968–976. [Google Scholar] [CrossRef]
- Burton, K.M.; Cao, H.; Chen, J.; Qiang, L.; Krueger, E.W.; Johnson, K.M.; Bamlet, W.R.; Zhang, L.; McNiven, M.A.; Razidlo, G.L. Dynamin 2 interacts with alpha-actinin 4 to drive tumor cell invasion. Mol. Biol. Cell 2020, 31, 439–451. [Google Scholar] [CrossRef]
- Mooren, O.L.; Kotova, T.I.; Moore, A.J.; Schafer, D.A. Dynamin2 GTPase and cortactin remodel actin filaments. J. Biol. Chem. 2009, 284, 23995–24005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppinga, R.D.; Krueger, E.W.; Weller, S.G.; Zhang, L.; Cao, H.; McNiven, M.A. Increased expression of the large GTPase dynamin 2 potentiates metastatic migration and invasion of pancreatic ductal carcinoma. Oncogene 2012, 31, 1228–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, H.M.; Sandle, J.; Mirza, T.A.; Li, N.F.; Hart, I.R. Ezrin interacts with cortactin to form podosomal rosettes in pancreatic cancer cells. Gut 2009, 58, 271–284. [Google Scholar] [CrossRef]
- Jeannot, P.; Nowosad, A.; Perchey, R.T.; Callot, C.; Bennana, E.; Katsube, T.; Mayeux, P.; Guillonneau, F.; Manenti, S.; Besson, A. p27(Kip1) promotes invadopodia turnover and invasion through the regulation of the PAK1/Cortactin pathway. eLife 2017, 6, e22207. [Google Scholar] [CrossRef]
- Moshfegh, Y.; Bravo-Cordero, J.J.; Miskolci, V.; Condeelis, J.; Hodgson, L. A Trio-Rac1-Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol. 2014, 16, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.T.; Cortesio, C.L.; Huttenlocher, A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 2009, 185, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goicoechea, S.M.; Zinn, A.; Awadia, S.S.; Snyder, K.; Garcia-Mata, R. A RhoG-mediated signaling pathway that modulates invadopodia dynamics in breast cancer cells. J. Cell Sci. 2017, 130, 1064–1077. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zhang, Y.; Kane, K.T.; Collins, M.A.; Simeone, D.M.; di Magliano, M.P.; Nguyen, K.T. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol. Cancer Res. 2015, 13, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Shields, M.A.; Dangi-Garimella, S.; Krantz, S.B.; Bentrem, D.J.; Munshi, H.G. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 2011, 286, 10495–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, A.; Matsumoto, S.; Yasumizu, Y.; Shojima, K.; Akama, T.; Eguchi, H.; Kikuchi, A. Localization of KRAS downstream target ARL4C to invasive pseudopods accelerates pancreatic cancer cell invasion. eLife 2021, 10, e66721. [Google Scholar] [CrossRef] [PubMed]
- Colombero, C.; Remy, D.; Antoine-Bally, S.; Mace, A.S.; Monteiro, P.; ElKhatib, N.; Fournier, M.; Dahmani, A.; Montaudon, E.; Montagnac, G.; et al. mTOR Repression in Response to Amino Acid Starvation Promotes ECM Degradation Through MT1-MMP Endocytosis Arrest. Adv. Sci. 2021, 8, e2101614. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zech, T.; McDonald, L.; Gonzalez, E.G.; Li, A.; Macpherson, I.; Schwarz, J.P.; Spence, H.; Futo, K.; Timpson, P.; et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J. Cell Biol. 2012, 199, 527–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Machesky, L.M. Cells assemble invadopodia-like structures and invade into matrigel in a matrix metalloprotease dependent manner in the circular invasion assay. PLoS ONE 2012, 7, e30605. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Cao, H.; Chen, J.; Weller, S.G.; Krueger, E.W.; Zhang, L.; Razidlo, G.L.; McNiven, M.A. Pancreatic tumor cell metastasis is restricted by MT1-MMP binding protein MTCBP-1. J. Cell Biol. 2019, 218, 317–332. [Google Scholar] [CrossRef] [Green Version]
- Kitano, A.; Shimasaki, T.; Chikano, Y.; Nakada, M.; Hirose, M.; Higashi, T.; Ishigaki, Y.; Endo, Y.; Takino, T.; Sato, H.; et al. Aberrant glycogen synthase kinase 3beta is involved in pancreatic cancer cell invasion and resistance to therapy. PLoS ONE 2013, 8, e55289. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Dai, L.; Niu, J. GPX2 silencing relieves epithelial-mesenchymal transition, invasion, and metastasis in pancreatic cancer by downregulating Wnt pathway. J. Cell Physiol. 2020, 235, 7780–7790. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, G.; Gao, F.; Liu, P.; Wang, H.; Tan, X. TSPAN1 upregulates MMP2 to promote pancreatic cancer cell migration and invasion via PLCgamma. Oncol. Rep. 2019, 41, 2117–2125. [Google Scholar] [CrossRef] [Green Version]
- Diaz, B.; Yuen, A.; Iizuka, S.; Higashiyama, S.; Courtneidge, S.A. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J. Cell Biol. 2013, 201, 279–292. [Google Scholar] [CrossRef]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, S.; Ren, H.; Sun, W.; Zhang, H.; Sun, J.; Yang, S.; Hao, J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014, 74, 2455–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhou, L.; Wei, B.; Qian, Z.; Wang, J.; Hui, H.; Sun, Y. miR1425p inhibits pancreatic cancer cell migration and invasion by targeting PIK3CA. Mol. Med. Rep. 2020, 22, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Matsuda, Y.; Kawahara, K.; Ishiwata, T.; Naito, Z. Secreted 70kDa lumican stimulates growth and inhibits invasion of human pancreatic cancer. Cancer Lett. 2012, 320, 31–39. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Uekita, T.; Hiraoka, N.; Fujii, S.; Kosuge, T.; Kanai, Y.; Nojima, Y.; Sakai, R. CUB domain-containing protein 1, a prognostic factor for human pancreatic cancers, promotes cell migration and extracellular matrix degradation. Cancer Res. 2010, 70, 5136–5146. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, T.A.; Lai, L.A.; Coleman, J.; Bronner, M.P.; Pan, S.; Chen, R. Arousal of cancer-associated stroma: Overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS ONE 2012, 7, e30219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Qiang, L.; Chen, J.; Johnson, K.M.; McNiven, M.A.; Razidlo, G.L. Synergistic metalloproteinase-based remodeling of matrix by pancreatic tumor and stromal cells. PLoS ONE 2021, 16, e0248111. [Google Scholar] [CrossRef]
- Goicoechea, S.M.; Garcia-Mata, R.; Staub, J.; Valdivia, A.; Sharek, L.; McCulloch, C.G.; Hwang, R.F.; Urrutia, R.; Yeh, J.J.; Kim, H.J.; et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene 2014, 33, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Eppinga, R.D.; Razidlo, G.L.; Krueger, E.W.; Chen, J.; Qiang, L.; McNiven, M.A. Stromal fibroblasts facilitate cancer cell invasion by a novel invadopodia-independent matrix degradation process. Oncogene 2016, 35, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Yoshitomi, H.; Takano, S.; Shida, T.; Kobayashi, S.; Ohtsuka, M.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Kato, A.; et al. FGF10/FGFR2 signal induces cell migration and invasion in pancreatic cancer. Br. J. Cancer 2008, 99, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Haage, A.; Schneider, I.C. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014, 28, 3589–3599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Moller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J. Cell Sci. 2007, 120, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef]
- Qian, B.; Wei, L.; Yang, Z.; He, Q.; Chen, H.; Wang, A.; Yang, D.; Li, Q.; Li, J.; Zheng, S.; et al. Hic-5 in pancreatic stellate cells affects proliferation, apoptosis, migration, invasion of pancreatic cancer cells and postoperative survival time of pancreatic cancer. Biomed. Pharmacother. 2020, 121, 109355. [Google Scholar] [CrossRef]
- Komura, T.; Sakai, Y.; Harada, K.; Kawaguchi, K.; Takabatake, H.; Kitagawa, H.; Wada, T.; Honda, M.; Ohta, T.; Nakanuma, Y.; et al. Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact. Cancer Sci. 2015, 106, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzing, C.; Lam, H.; Tsang, C.M.; Rimmer, A.; Arroyo-Berdugo, Y.; Calle, Y.; Wells, C.M. TIMP-2 secreted by monocyte-like cells is a potent suppressor of invadopodia formation in pancreatic cancer cells. BMC Cancer 2019, 19, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Razidlo, G.L.; Burton, K.M.; McNiven, M.A. Interleukin-6 promotes pancreatic cancer cell migration by rapidly activating the small GTPase CDC42. J. Biol. Chem. 2018, 293, 11143–11153. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Wei, W. RAB5A promotes the formation of filopodia in pancreatic cancer cells via the activation of cdc42 and beta1-integrin. Biochem. Biophys. Res. Commun. 2021, 535, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Razidlo, G.L.; Wang, Y.; Chen, J.; Krueger, E.W.; Billadeau, D.D.; McNiven, M.A. Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev. Cell 2013, 24, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Welsch, T.; Endlich, K.; Giese, T.; Buchler, M.W.; Schmidt, J. Eps8 is increased in pancreatic cancer and required for dynamic actin-based cell protrusions and intercellular cytoskeletal organization. Cancer Lett. 2007, 255, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Yokotani, K.; Saibara, T. BART inhibits pancreatic cancer cell invasion by Rac1 inactivation through direct binding to active Rac1. Neoplasia 2012, 14, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Wang, S.; Gunther, L.K.; Kitajiri, S.; Li, C.; Sakamoto, T. The actin-bundling protein TRIOBP-4 and -5 promotes the motility of pancreatic cancer cells. Cancer Lett. 2015, 356, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Yoshida, H.; Katori, M.; Migita, T.; Muramatsu, Y.; Miyake, M.; Ishikawa, Y.; Saiura, A.; Iemura, S.I.; Natsume, T.; et al. Tankyrase-Binding Protein TNKS1BP1 Regulates Actin Cytoskeleton Rearrangement and Cancer Cell Invasion. Cancer Res. 2017, 77, 2328–2338. [Google Scholar] [CrossRef] [Green Version]
- Bizzozero, L.; Pergolizzi, M.; Pascal, D.; Maldi, E.; Villari, G.; Erriquez, J.; Volante, M.; Serini, G.; Marchio, C.; Bussolino, F.; et al. Tumoral Neuroligin 1 Promotes Cancer-Nerve Interactions and Synergizes with the Glial Cell Line-Derived Neurotrophic Factor. Cells 2022, 11, 280. [Google Scholar] [CrossRef]
- Broussard, J.A.; Webb, D.J.; Kaverina, I. Asymmetric focal adhesion disassembly in motile cells. Curr. Opin. Cell Biol. 2008, 20, 85–90. [Google Scholar] [CrossRef]
- Maziveyi, M.; Alahari, S.K. Cell matrix adhesions in cancer: The proteins that form the glue. Oncotarget 2017, 8, 48471–48487. [Google Scholar] [CrossRef] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Kanteti, R.; Batra, S.K.; Lennon, F.E.; Salgia, R. FAK and paxillin, two potential targets in pancreatic cancer. Oncotarget 2016, 7, 31586–31601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311. [Google Scholar] [CrossRef]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—The update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Layseca, P.; Icha, J.; Hamidi, H.; Ivaska, J. Integrin trafficking in cells and tissues. Nat. Cell Biol. 2019, 21, 122–132. [Google Scholar] [CrossRef]
- Li, N.F.; Gemenetzidis, E.; Marshall, F.J.; Davies, D.; Yu, Y.; Frese, K.; Froeling, F.E.; Woolf, A.K.; Feakins, R.M.; Naito, Y.; et al. RhoC interacts with integrin alpha5beta1 and enhances its trafficking in migrating pancreatic carcinoma cells. PLoS ONE 2013, 8, e81575. [Google Scholar] [CrossRef]
- He, R.; Wang, M.; Zhao, C.; Shen, M.; Yu, Y.; He, L.; Zhao, Y.; Chen, H.; Shi, X.; Zhou, M.; et al. TFEB-driven autophagy potentiates TGF-beta induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 340. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004, 23, 1448–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Wang, H.; Manavathi, B.; Fok, J.Y.; Mann, A.P.; Kumar, R.; Mehta, K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006, 66, 10525–10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Z.; Kunnumakkara, A.B.; Wang, H.; Matsuo, Y.; Diagaradjane, P.; Harikumar, K.B.; Ramachandran, V.; Sung, B.; Chakraborty, A.; Bresalier, R.S.; et al. Neutrophil gelatinase-associated lipocalin: A novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 2008, 68, 6100–6108. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, S.J.; Min, H.J.; Jo, J.Y.; Park, E.H.; Koh, S.S. PAUF promotes adhesiveness of pancreatic cancer cells by modulating focal adhesion kinase. Exp. Mol. Med. 2011, 43, 291–297. [Google Scholar] [CrossRef]
- Yue, S.; Mu, W.; Zoller, M. Tspan8 and CD151 promote metastasis by distinct mechanisms. Eur. J. Cancer 2013, 49, 2934–2948. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Guha, S.; Wang, H.; Fok, J.Y.; Koul, D.; Abbruzzese, J.; Mehta, K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 1997–2005. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Li, H.; Li, N.; Sun, H.; Li, Q.; Shen, X. WNT5A/JNK signaling regulates pancreatic cancer cells migration by Phosphorylating Paxillin. Pancreatology 2013, 13, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, J.; Zhang, Y.; Zhou, Z.; Cui, X.; Zhang, L.; Fung, K.M.; Zheng, W.; Allard, F.D.; Yee, E.U.; et al. ZIP4 Promotes Pancreatic Cancer Progression by Repressing ZO-1 and Claudin-1 through a ZEB1-Dependent Transcriptional Mechanism. Clin. Cancer Res. 2018, 24, 3186–3196. [Google Scholar] [CrossRef] [Green Version]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Syed, N.; Therachiyil, L.; Nisar, S.; Hashem, S.; Macha, M.A.; Yadav, S.K.; Krishnankutty, R.; Muralitharan, S.; Al-Naemi, H.; et al. Claudin-1, A Double-Edged Sword in Cancer. Int. J. Mol. Sci. 2020, 21, 569. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, Y.; Yang, J.; Zhan, H.; Zhou, Z.; Jiang, Y.; Shi, X.; Fan, X.; Zhang, J.; Luo, W.; et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology 2021, 160, 1771–1783.e1. [Google Scholar] [CrossRef] [PubMed]
- Mu, G.; Ding, Q.; Li, H.; Zhang, L.; Zhang, L.; He, K.; Wu, L.; Deng, Y.; Yang, D.; Wu, L.; et al. Gastrin stimulates pancreatic cancer cell directional migration by activating the Galpha12/13-RhoA-ROCK signaling pathway. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.P.; Hamory, M.W.; Verderame, M.F.; Zagon, I.S. Quantitative analysis of gastrin mRNA and peptide in normal and cancerous human pancreas. Int. J. Mol. Med. 1998, 2, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Shih, A.; Wu, Y.; McLaughlin, P.J.; Zagon, I.S. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am. J. Physiol. 1996, 270, R1078–R1084. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Horwitz, A.R. Adhesion dynamics at a glance. J. Cell Sci. 2011, 124, 3923–3927. [Google Scholar] [CrossRef] [Green Version]
- Nogales, E. Structural insights into microtubule function. Annu. Rev. Biochem. 2000, 69, 277–302. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Microtubules at focal adhesions—A double-edged sword. J. Cell Sci. 2019, 132, jcs.232843. [Google Scholar] [CrossRef] [Green Version]
- Stehbens, S.; Wittmann, T. Targeting and transport: How microtubules control focal adhesion dynamics. J. Cell Biol. 2012, 198, 481–489. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef]
- Tanabe, K.; Takei, K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J. Cell Biol. 2009, 185, 939–948. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, H.; Chen, J.; McNiven, M.A. A direct interaction between the large GTPase dynamin-2 and FAK regulates focal adhesion dynamics in response to active Src. Mol. Biol. Cell 2011, 22, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Dallas, M.R.; Chen, S.H.; Streppel, M.M.; Sharma, S.; Maitra, A.; Konstantopoulos, K. Sialofucosylated podocalyxin is a functional E- and L-selectin ligand expressed by metastatic pancreatic cancer cells. Am. J. Physiol. Cell Physiol. 2012, 303, C616–C624. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Furihata, M.; Naganuma, S.; Dabanaka, K.; Hanazaki, K.; Saibara, T. Podocalyxin-like protein, linked to poor prognosis of pancreatic cancers, promotes cell invasion by binding to gelsolin. Cancer Sci. 2016, 107, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Chijiiwa, Y.; Moriyama, T.; Ohuchida, K.; Nabae, T.; Ohtsuka, T.; Miyasaka, Y.; Fujita, H.; Maeyama, R.; Manabe, T.; Abe, A.; et al. Overexpression of microRNA-5100 decreases the aggressive phenotype of pancreatic cancer cells by targeting PODXL. Int. J. Oncol. 2016, 48, 1688–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.S.; Shea, D.J.; Mistriotis, P.; Tuntithavornwat, S.; Law, R.A.; Bieber, J.M.; Zheng, L.; Konstantopoulos, K. A Direct Podocalyxin-Dynamin-2 Interaction Regulates Cytoskeletal Dynamics to Promote Migration and Metastasis in Pancreatic Cancer Cells. Cancer Res. 2019, 79, 2878–2891. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Wang, J.; Xu, J.; Jiang, X.; Liu, X.; Jiang, J. Dynamic regulation of KIF15 phosphorylation and acetylation promotes focal adhesions disassembly in pancreatic cancer. Cell Death Dis. 2022, 13, 896. [Google Scholar] [CrossRef]
- Tuntithavornwat, S.; Shea, D.J.; Wong, B.S.; Guardia, T.; Lee, S.J.; Yankaskas, C.L.; Zheng, L.; Kontrogianni-Konstantopoulos, A.; Konstantopoulos, K. Giant obscurin regulates migration and metastasis via RhoA-dependent cytoskeletal remodeling in pancreatic cancer. Cancer Lett. 2022, 526, 155–167. [Google Scholar] [CrossRef]
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Callaway, M.K.; Rodriguez-Merced, N.J.; Crampton, A.L.; Carlson, M.; Emme, K.B.; Ensminger, E.A.; Kinne, A.A.; Schrope, J.H.; Rasmussen, H.R.; et al. Stromal architecture directs early dissemination in pancreatic ductal adenocarcinoma. JCI Insight 2022, 7, e150330. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, V.B.; Gutierrez Ruiz, O.L.; Razidlo, G.L. The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights. Cancers 2023, 15, 2169. https://doi.org/10.3390/cancers15072169
Joshi VB, Gutierrez Ruiz OL, Razidlo GL. The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights. Cancers. 2023; 15(7):2169. https://doi.org/10.3390/cancers15072169
Chicago/Turabian StyleJoshi, Vidhu B., Omar L. Gutierrez Ruiz, and Gina L. Razidlo. 2023. "The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights" Cancers 15, no. 7: 2169. https://doi.org/10.3390/cancers15072169
APA StyleJoshi, V. B., Gutierrez Ruiz, O. L., & Razidlo, G. L. (2023). The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights. Cancers, 15(7), 2169. https://doi.org/10.3390/cancers15072169