
Reviewing the Prospective Pharmacological Potential of Isothiocyanates in Fight against Female-Specific Cancers

 , ,
, ,  , , ,
, , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. General Characteristics, Sources, and Biological Importance of Isothiocyanates
3. Impacts of Isothiocyanates on Female-Specific Cancers
3.1. Impacts of Isothiocyanates on Cancer Proliferation, Migration, and Invasion-Related Signal Transduction Pathways
3.1.1. Impacts of SFN on Cancer Proliferation, Migration, and Invasion-Related Signal Transduction Pathways
3.1.2. Impacts of BITC on Cancer Proliferation, Migration, and Invasion-Related Signal Transduction Pathways
3.1.3. Impacts of PEITC on Cancer Proliferation, Migration, and Invasion-Related Signal Transduction Pathways
3.2. Impacts of Isothiocyanates on Modulation of Cell Cycle
3.2.1. Impacts of SFN on Modulation of Cell Cycle
3.2.2. Impacts of BITC on Modulation of Cell Cycle
3.2.3. Impacts of PEITC on Modulation of Cell Cycle
3.3. Impacts of Isothiocyanates on Apoptosis
3.3.1. Impacts of SFN on Apoptosis
3.3.2. Impacts of BITC on Apoptosis
3.3.3. Impacts of PEITC on Apoptosis
3.4. Impacts of Isothiocyanates on Modulation of Autophagy
3.4.1. Impacts of SFN on Modulation of Autophagy
3.4.2. Impacts of BITC on Modulation of Autophagy
3.4.3. Impacts of PEITC on Modulation of Autophagy
3.5. Impacts of Isothiocyanates on Cancer Stem Cells (CSCs)
3.5.1. Impacts of SFN on CSCs
3.5.2. Impacts of BITC on CSCs
3.5.3. Impacts of PEITC on CSCs
4. Combinatorial Studies on ITCs and Anticancer Drugs/Phytochemicals
4.1. Combinatorial Treatment of SFN and Anticancer Drugs/Phytochemicals
4.2. Combinatorial Treatment of BITC and Anticancer Drugs/Phytochemicals
4.3. Combinatorial Treatment of PEITC and Anticancer Drugs/Phytochemicals
5. Isothiocyanates and Their Anticancer Potential in Animal Model
5.1. SFN and Their Anticancer Potential in Animal Model
5.2. BITC and Their Anticancer Potential in Animal Model
5.3. PEITC and Their Anticancer Potential in Animal Model
6. Isothiocyanates in Clinical Trials
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP1 | activating protein-1 |
| AMPK | AMP-activated protein kinase |
| Bcl-2 | B-cell leukemia/lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| BITC | benzyl isothiocyanate |
| CDH1 | cadherin 1 |
| CDK | cyclin-dependent kinase |
| DAPK1 | death-associated protein kinase 1 |
| ECM | extracellular matrix |
| EGCG | epigallocatechin gallate |
| ERK | extracellular signal-regulated kinase |
| FOX1 | forkhead box O1 |
| GLUT-1 | glucose transporter-1 |
| GRP78 | glucose-regulating protein 78 |
| GSK3-β | glycogen synthase kinase 3-β |
| GSTP1 | glutathione S-transferase Pi |
| HIF-1 | hypoxia-inducible factor-1 |
| HPV | human papilloma virus |
| ICAM1 | intercellular adhesion molecule |
| INK4 | inhibitors of CDK4 |
| JNK | Jun N-terminal Kinase |
| KLF4 | kruppel-like factor 4 |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor kappa B |
| NOD/SCID | nonobese diabetic/severe combined immunodeficiency |
| Nrf-2 | nuclear factor erythroid 2–related factor 2 |
| Oct-4 | octamer-binding transcription factor 4 |
| PEITC | phenethyl isothiocyanate |
| PERK | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PI3K | phosphatidyl inositol 3 kinase |
| RARβ | retinoic acid receptor β |
| ROS | reactive oxygen species |
| SFN | sulforaphane |
| SMAD | suppressor of mothers against decapentaplegic |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor |
| TNBCs | triple-negative breast cancer cells |
| VEGF | vascular endothelial growth factor |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Marchbanks, P.A.; McDonald, J.A.; Wilson, H.G.; Folger, S.G.; Mandel, M.G.; Daling, J.R.; Bernstein, L.; Malone, K.E.; Ursin, G.; Strom, B.L.; et al. Oral contraceptives and the risk of breast cancer. N. Engl. J. Med. 2002, 346, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Colditz, G.A.; Rosner, B. Cumulative risk of breast cancer to age 70 years according to risk factor status: Data from the Nurses’ Health Study. Am. J. Epidemiol. 2000, 152, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Willett, W.C.; Rosner, B.A.; Hankinson, S.E.; Michels, K.B. Cigarette smoking and the incidence of breast cancer. Arch. Intern. Med. 2011, 171, 125–133. [Google Scholar] [CrossRef]
- Niraula, S.; Ocana, A.; Ennis, M.; Goodwin, P.J. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: A meta-analysis. Breast Cancer Res. Treat. 2012, 134, 769–781. [Google Scholar] [CrossRef]
- Estebanez, N.; Gomez-Acebo, I.; Palazuelos, C.; Llorca, J.; Dierssen-Sotos, T. Vitamin D exposure and Risk of Breast Cancer: A meta-analysis. Sci. Rep. 2018, 8, 9039. [Google Scholar] [CrossRef]
- Elme, A.; Utriainen, M.; Kellokumpu-Lehtinen, P.; Palva, T.; Luoto, R.; Nikander, R.; Huovinen, R.; Kautiainen, H.; Jarvenpaa, S.; Penttinen, H.M.; et al. Obesity and physical inactivity are related to impaired physical health of breast cancer survivors. Anticancer Res. 2013, 33, 1595–1602. [Google Scholar]
- Parise, C.; Caggiano, V. The influence of marital status and race/ethnicity on risk of mortality for triple negative breast cancer. PLoS ONE 2018, 13, e0196134. [Google Scholar] [CrossRef]
- Wang, J.; John, E.M.; Horn-Ross, P.L.; Ingles, S.A. Dietary fat, cooking fat, and breast cancer risk in a multiethnic population. Nutr. Cancer 2008, 60, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, C.; Lauby-Secretan, B.; Bello, P.Y.; Chajes, V.; Romieu, I. Female breast cancer and alcohol consumption: A review of the literature. Am. J. Prev. Med. 2014, 46, S16–S25. [Google Scholar] [CrossRef] [PubMed]
- Brüggmann, D.; Quinkert-Schmolke, K.; Jaque, J.M.; Quarcoo, D.; Bohlmann, M.K.; Klingelhöfer, D.; Groneberg, D.A. Global cervical cancer research: A scientometric density equalizing mapping and socioeconomic analysis. PLoS ONE 2022, 17, e0261503. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. S5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Clavel, C.; Masure, M.; Bory, J.P.; Putaud, I.; Mangeonjean, C.; Lorenzato, M.; Nazeyrollas, P.; Gabriel, R.; Quereux, C.; Birembaut, P. Human papillomavirus testing in primary screening for the detection of high-grade cervical lesions: A study of 7932 women. Br. J. Cancer 2001, 84, 1616–1623. [Google Scholar] [CrossRef]
- Hillemanns, P.; Soergel, P.; Hertel, H.; Jentschke, M. Epidemiology and Early Detection of Cervical Cancer. Oncol. Res. Treat. 2016, 39, 501–506. [Google Scholar] [CrossRef]
- Moreno, V.; Bosch, F.X.; Munoz, N.; Meijer, C.J.; Shah, K.V.; Walboomers, J.M.; Herrero, R.; Franceschi, S.; International Agency for Research on Cancer. Multicentric Cervical Cancer Study Group. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092. [Google Scholar] [CrossRef]
- Schrager, S.; Potter, B.E. Diethylstilbestrol exposure. Am. Fam. Physician 2004, 69, 2395–2400. [Google Scholar]
- Louie, K.S.; Castellsague, X.; de Sanjose, S.; Herrero, R.; Meijer, C.J.; Shah, K.; Munoz, N.; Bosch, F.X.; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Smoking and passive smoking in cervical cancer risk: Pooled analysis of couples from the IARC multicentric case-control studies. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, N.M.; Bin Judia, S.S.; Mishra, K.N.; Shoukri, M.M.; Alsbeih, G.A. Genetic Predisposition to Cervical Cancer and the Association With XRCC1 and TGFB1 Polymorphisms. Int. J. Gynecol. Cancer 2017, 27, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Tindle, R.W. Immune evasion in human papillomavirus-associated cervical cancer. Nat. Rev. Cancer 2002, 2, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Maner, S.; Betz, R.; Angstrom, T.; Stendahl, U.; Bergman, F.; Zetterberg, A.; Wallin, K.L. Genetic alterations in cervical carcinomas: Frequent low-level amplifications of oncogenes are associated with human papillomavirus infection. Int. J. Cancer 2002, 101, 427–433. [Google Scholar] [CrossRef]
- Baay, M.F.; Kjetland, E.F.; Ndhlovu, P.D.; Deschoolmeester, V.; Mduluza, T.; Gomo, E.; Friis, H.; Midzi, N.; Gwanzura, L.; Mason, P.R.; et al. Human papillomavirus in a rural community in Zimbabwe: The impact of HIV co-infection on HPV genotype distribution. J. Med. Virol. 2004, 73, 481–485. [Google Scholar] [CrossRef]
- Bellaminutti, S.; Seraceni, S.; De Seta, F.; Gheit, T.; Tommasino, M.; Comar, M. HPV and Chlamydia trachomatis co-detection in young asymptomatic women from high incidence area for cervical cancer. J. Med. Virol. 2014, 86, 1920–1925. [Google Scholar] [CrossRef]
- Ziegler, R.G.; Weinstein, S.J.; Fears, T.R. Nutritional and genetic inefficiencies in one-carbon metabolism and cervical cancer risk. J. Nutr 2002, 132, 2345S–2349S. [Google Scholar] [CrossRef]
- Parikh, S.; Brennan, P.; Boffetta, P. Meta-analysis of social inequality and the risk of cervical cancer. Int. J. Cancer 2003, 105, 687–691. [Google Scholar] [CrossRef]
- Sabeena, S.; Bhat, P.; Kamath, V.; Arunkumar, G. Possible non-sexual modes of transmission of human papilloma virus. J. Obstet. Gynaecol. Res. 2017, 43, 429–435. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef]
- Cabasag, C.J.; Fagan, P.J.; Ferlay, J.; Vignat, J.; Laversanne, M.; Liu, L.; van der Aa, M.A.; Bray, F.; Soerjomataram, I. Ovarian cancer today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020. Int. J. Cancer 2022, 151, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Gilks, C.B.; Ionescu, D.N.; Kalloger, S.E.; Kobel, M.; Irving, J.; Clarke, B.; Santos, J.; Le, N.; Moravan, V.; Swenerton, K.; et al. Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Hum. Pathol. 2008, 39, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.; Fuh, K. Recent Advances in Understanding, Diagnosing, and Treating Ovarian Cancer. F1000Res 2017, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, A.; Quinn, M.; Thigpen, T.; Vermorken, J.; Avall-Lundqvist, E.; Bookman, M.; Bowtell, D.; Brady, M.; Casado, A.; Cervantes, A.; et al. 2004 consensus statements on the management of ovarian cancer: Final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004). Ann. Oncol. 2005, 16 (Suppl. S8), viii7–viii12. [Google Scholar] [CrossRef]
- Crijns, A.P.; Fehrmann, R.S.; de Jong, S.; Gerbens, F.; Meersma, G.J.; Klip, H.G.; Hollema, H.; Hofstra, R.M.; te Meerman, G.J.; de Vries, E.G.; et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009, 6, e24. [Google Scholar] [CrossRef]
- Alam, S.S.M.; Uddin, F.; Khan, F.B.; Kamal, M.A.; Hoque, M.J.P.P. Therapeutic and pharmacological potential of tanshinones against lung cancer: A systematic review. Phytomed. Plus 2021, 2, 100202. [Google Scholar] [CrossRef]
- Hussain, Y.; Alsharif, K.F.; Aschner, M.; Theyab, A.; Khan, F.; Saso, L.; Khan, H. Therapeutic Role of Carotenoids in Blood Cancer: Mechanistic Insights and Therapeutic Potential. Nutrients 2022, 14, 1949. [Google Scholar] [CrossRef]
- Ansari, M.A.; Khan, F.B.; Safdari, H.A.; Almatroudi, A.; Alzohairy, M.A.; Safdari, M.; Amirizadeh, M.; Rehman, S.; Equbal, M.J.; Hoque, M. Prospective therapeutic potential of Tanshinone IIA: An updated overview. Pharmacol. Res. 2021, 164, 105364. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Orsulic, S.; Ashok, B.T. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol. Cancer Ther. 2007, 6, 334–345. [Google Scholar] [CrossRef]
- Khan, F.B.; Ansari, M.A.; Uddin, S.; Palakott, A.R.; Anwar, I.; Almatroudi, A.; Alomary, M.N.; Alrumaihi, F.; Aba Alkhayl, F.F.; Alghamdi, S. Prospective Role of Bioactive Molecules and Exosomes in the Therapeutic Potential of Camel Milk against Human Diseases: An Updated Perspective. Life 2022, 12, 990. [Google Scholar] [CrossRef]
- Khan, F.B.; Singh, P.; Jamous, Y.F.; Ali, S.A.; Uddin, S.; Zia, Q.; Jena, M.K.; Khan, M.; Owais, M.; Huang, C.Y. Multifaceted Pharmacological Potentials of Curcumin, Genistein, and Tanshinone IIA through Proteomic Approaches: An In-Depth Review. Cancers 2023, 15, 249. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.; Khan, F.B.; Akhtar, S.; Ahmad, A.; Uddin, S. The plasticity of pancreatic cancer stem cells: Implications in therapeutic resistance. Cancer Metastasis Rev. 2021, 40, 691–720. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, M.; Quispe, C.; Sharifi-Rad, J.; Pons-Fuster, E.; Lopez-Jornet, P.; Zam, W.; Das, T.; Dey, A.; Kumar, M.; Pentea, M.; et al. Naturally-Occurring Bioactives in Oral Cancer: Preclinical and Clinical Studies, Bottlenecks and Future Directions. Front. Biosci. (Sch. Ed.) 2022, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, M.; Quispe, C.; Koirala, N.; Khadka, S.; Salgado-Castillo, C.M.; Akram, M.; Anum, R.; Yeskaliyeva, B.; Cruz-Martins, N.; Martorell, M.; et al. Bioactive Effects of Curcumin in Human Immunodeficiency Virus Infection Along with the Most Effective Isolation Techniques and Type of Nanoformulations. Int. J. Nanomed. 2022, 17, 3619–3632. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, M.; Quispe, C.; Herrera-Bravo, J.; Fernández-Ochoa, Á.; Emamzadeh-Yazdi, S.; Adetunji, C.O.; Memudu, A.E.; Otlewska, A.; Bogdan, P.; Antolak, H.; et al. A Review on Tradescantia: Phytochemical Constituents, Biological Activities and Health-Promoting Effects. Front. Biosci. (Landmark Ed.) 2022, 27, 197. [Google Scholar] [CrossRef]
- Javed, Z.; Sadia, H.; Iqbal, M.J.; Shamas, S.; Malik, K.; Ahmed, R.; Raza, S.; Butnariu, M.; Cruz-Martins, N.; Sharifi-Rad, J. Apigenin role as cell-signaling pathways modulator: Implications in cancer prevention and treatment. Cancer Cell. Int. 2021, 21, 189. [Google Scholar] [CrossRef]
- Gasmi, A.; Peana, M.; Arshad, M.; Butnariu, M.; Menzel, A.; Bjørklund, G. Krebs cycle: Activators, inhibitors and their roles in the modulation of carcinogenesis. Arch. Toxicol. 2021, 95, 1161–1178. [Google Scholar] [CrossRef]
- Chiang, J.T.; Badrealam, K.F.; Shibu, M.A.; Kuo, C.H.; Huang, C.Y.; Chen, B.C.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.-Y. Eriobotrya japonica ameliorates cardiac hypertrophy in H9c2 cardiomyoblast and in spontaneously hypertensive rats. Environ. Toxicol. 2018, 33, 1113–1122. [Google Scholar] [CrossRef]
- Khan, B.F.; Dwivedi, S.; Konwar, R.; Zubair, S.; Owais, M. Potential of bacterial culture media in biofabrication of metal nanoparticles and the therapeutic potential of the as-synthesized nanoparticles in conjunction with artemisinin against MDA-MB-231 breast cancer cells. J. Cell. Physiol. 2019, 234, 6951–6964. [Google Scholar] [CrossRef]
- Kung, Y.L.; Lu, C.Y.; Badrealam, K.F.; Kuo, W.W.; Shibu, M.A.; Day, C.H.; Chen, R.J.; Lu, S.Y.; Padma, V.V.; Huang, C.Y. Cardioprotective potential of amygdalin against angiotensin II induced cardiac hypertrophy, oxidative stress and inflammatory responses through modulation of Nrf2 and NF-κB activation. Environ. Toxicol. 2021, 36, 926–934. [Google Scholar] [CrossRef]
- Lin, Y.-M.; Badrealam, K.F.; Kuo, W.-W.; Lai, P.F.; Chen, W.S.-T.; Day, C.H.; Ho, T.-J.; Viswanadha, V.P.; Shibu, M.A.; Huang, C.-Y. Nerolidol improves cardiac function in spontaneously hypertensive rats by inhibiting cardiac inflammation and remodelling associated TLR4/NF-κB signalling cascade. Food Chem. Toxicol. 2021, 147, 111837. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-M.; Badrealam, K.F.; Kuo, C.-H.; Daddam, J.; Shibu, M.A.; Lin, K.-H.; Ho, T.-J.; Viswanadha, V.P.; Kuo, W.-W.; Huang, C.-Y. Small Molecule Compound Nerolidol attenuates Hypertension induced hypertrophy in spontaneously hypertensive rats through modulation of Mel-18-IGF-IIR signalling. Phytomedicine 2021, 84, 153450. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, B.; Santolamazza, C.; Simonelli, F.; Rubattu, S. Phytochemical Compounds and Protection from Cardiovascular Diseases: A State of the Art. BioMed Res. Int. 2015, 2015, 918069. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed]
- Pohl, F.; Kong Thoo Lin, P. The Potential Use of Plant Natural Products and Plant Extracts with Antioxidant Properties for the Prevention/Treatment of Neurodegenerative Diseases: In Vitro, In Vivo and Clinical Trials. Molecules 2018, 23, 3283. [Google Scholar] [CrossRef]
- Wang, S.-H.; Wu, H.-C.; Badrealam, K.F.; Kuo, Y.-H.; Chao, Y.-P.; Hsu, H.-H.; Bau, D.-T.; Viswanadha, V.P.; Chen, Y.-H.; Lio, P.-J.; et al. Taiwanin E induces cell cycle arrest and apoptosis in arecoline/4-NQO-induced oral cancer cells through modulation of the ERK signaling pathway. Front. Oncol. 2019, 9, 1309. [Google Scholar] [CrossRef]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef]
- Fischer, N.; Seo, E.J.; Efferth, T. Prevention from radiation damage by natural products. Phytomedicine 2018, 47, 192–200. [Google Scholar] [CrossRef]
- Banudevi, S.; Swaminathan, S.; Maheswari, K.U. Pleiotropic Role of Dietary Phytochemicals in Cancer: Emerging Perspectives for Combinational Therapy. Nutr. Cancer 2015, 67, 1021–1048. [Google Scholar] [CrossRef]
- Pezzani, R.; Salehi, B.; Vitalini, S.; Iriti, M.; Zuniga, F.A.; Sharifi-Rad, J.; Martorell, M.; Martins, N. Synergistic Effects of Plant Derivatives and Conventional Chemotherapeutic Agents: An Update on the Cancer Perspective. Medicina 2019, 55, 110. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Palliyaguru, D.L.; Yuan, J.M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the Power of Plants to People. Mol. Nutr. Food Res. 2018, 62, e1700965. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Hampton, M.B. Biological targets of isothiocyanates. Biochim. Biophys. Acta 2011, 1810, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Murillo, G.; Mehta, R.G. Cruciferous vegetables and cancer prevention. Nutr. Cancer 2001, 41, 17–28. [Google Scholar] [CrossRef]
- Hecht, S.S. Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 2000, 32, 395–411. [Google Scholar] [CrossRef]
- Fimognari, C.; Lenzi, M.; Hrelia, P. Chemoprevention of cancer by isothiocyanates and anthocyanins: Mechanisms of action and structure-activity relationship. Curr. Med. Chem. 2008, 15, 440–447. [Google Scholar] [CrossRef]
- Jacob, C. A scent of therapy: Pharmacological implications of natural products containing redox-active sulfur atoms. Nat. Prod. Rep. 2006, 23, 851–863. [Google Scholar] [CrossRef]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanisms of action of isothiocyanates in cancer chemoprevention: An update. Food Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef]
- Shoaib, S.; Ansari, M.A.; Ghazwani, M.; Hani, U.; Jamous, Y.F.; Alali, Z.; Wahab, S.; Ahmad, W.; Weir, S.A.; Alomary, M.N. Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers 2023, 15, 697. [Google Scholar] [CrossRef]
- Pawlik, A.; Wiczk, A.; Kaczynska, A.; Antosiewicz, J.; Herman-Antosiewicz, A. Sulforaphane inhibits growth of phenotypically different breast cancer cells. Eur. J. Nutr. 2013, 52, 1949–1958. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.C.; Singletary, K. Regulation of estrogen receptor alpha expression in human breast cancer cells by sulforaphane. J. Nutr. Biochem. 2009, 20, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kanematsu, S.; Uehara, N.; Miki, H.; Yoshizawa, K.; Kawanaka, A.; Yuri, T.; Tsubura, A. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010, 30, 3381–3390. [Google Scholar] [PubMed]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Korkaya, H.; Liu, S.; Lee, H.F.; Newman, B.; Yu, Y.; Clouthier, S.G.; Schwartz, S.J.; Wicha, M.S.; et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin. Cancer Res. 2010, 16, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Adamczyk-Grochala, J.; Deregowska, A.; Wnuk, M. Sulforaphane-Induced Cell Cycle Arrest and Senescence are accompanied by DNA Hypomethylation and Changes in microRNA Profile in Breast Cancer Cells. Theranostics 2017, 7, 3461–3477. [Google Scholar] [CrossRef]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018, 143, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Noh, E.M.; Han, J.H.; Kim, J.M.; Hwang, B.M.; Kim, B.S.; Lee, S.H.; Jung, S.H.; Youn, H.J.; Chung, E.Y.; et al. Sulforaphane controls TPA-induced MMP-9 expression through the NF-kappaB signaling pathway, but not AP-1, in MCF-7 breast cancer cells. BMB Rep. 2013, 46, 201–206. [Google Scholar] [CrossRef]
- Yang, F.; Wang, F.; Liu, Y.; Wang, S.; Li, X.; Huang, Y.; Xia, Y.; Cao, C. Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells. Life Sci. 2018, 213, 149–157. [Google Scholar] [CrossRef]
- Kan, S.F.; Wang, J.; Sun, G.X. Sulforaphane regulates apoptosis- and proliferation-related signaling pathways and synergizes with cisplatin to suppress human ovarian cancer. Int. J. Mol. Med. 2018, 42, 2447–2458. [Google Scholar] [CrossRef]
- Chang, C.C.; Hung, C.M.; Yang, Y.R.; Lee, M.J.; Hsu, Y.C. Sulforaphane induced cell cycle arrest in the G2/M phase via the blockade of cyclin B1/CDC2 in human ovarian cancer cells. J. Ovarian Res. 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.S.; Kumar, S.; Chamala, S.; Shah, J.; Pal, J.; Haider, M.; Seward, S.; Qazi, A.M.; Morris, R.; Semaan, A.; et al. Sulforaphane induces cell cycle arrest by protecting RB-E2F-1 complex in epithelial ovarian cancer cells. Mol. Cancer 2010, 9, 47. [Google Scholar] [CrossRef]
- Pastorek, M.; Simko, V.; Takacova, M.; Barathova, M.; Bartosova, M.; Hunakova, L.; Sedlakova, O.; Hudecova, S.; Krizanova, O.; Dequiedt, F.; et al. Sulforaphane reduces molecular response to hypoxia in ovarian tumor cells independently of their resistance to chemotherapy. Int. J. Oncol. 2015, 47, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.M.; Tsai, C.C.; Hsu, Y.C. Sulforaphane, a Dietary Isothiocyanate, Induces G(2)/M Arrest in Cervical Cancer Cells through CyclinB1 Downregulation and GADD45beta/CDC2 Association. Int. J. Mol. Sci. 2016, 17, 1530. [Google Scholar] [CrossRef] [PubMed]
- Ali Khan, M.; Kedhari Sundaram, M.; Hamza, A.; Quraishi, U.; Gunasekera, D.; Ramesh, L.; Goala, P.; Al Alami, U.; Ansari, M.Z.; Rizvi, T.A.; et al. Sulforaphane Reverses the Expression of Various Tumor Suppressor Genes by Targeting DNMT3B and HDAC1 in Human Cervical Cancer Cells. Evidence-Based Complement. Altern. Med. 2015, 2015, 412149. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, G.Y.; Bae, S.J.; Yoo, Y.H.; Choi, Y.H. Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol. Rep. 2007, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Satyan, K.S.; Swamy, N.; Dizon, D.S.; Singh, R.; Granai, C.O.; Brard, L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: Role of caspase and MAPK activation. Gynecol. Oncol. 2006, 103, 261–270. [Google Scholar] [CrossRef]
- Xiao, D.; Vogel, V.; Singh, S.V. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol. Cancer Ther. 2006, 5, 2931–2945. [Google Scholar] [CrossRef]
- Sehrawat, A.; Singh, S.V. Benzyl isothiocyanate inhibits epithelial-mesenchymal transition in cultured and xenografted human breast cancer cells. Cancer Prev. Res. 2011, 4, 1107–1117. [Google Scholar] [CrossRef]
- Sehrawat, A.; Kim, S.H.; Vogt, A.; Singh, S.V. Suppression of FOXQ1 in benzyl isothiocyanate-mediated inhibition of epithelial-mesenchymal transition in human breast cancer cells. Carcinogenesis 2013, 34, 864–873. [Google Scholar] [CrossRef]
- Xiao, D.; Bommareddy, A.; Kim, S.H.; Sehrawat, A.; Hahm, E.R.; Singh, S.V. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE 2012, 7, e32597. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sehrawat, A.; Singh, S.V. Dietary chemopreventative benzyl isothiocyanate inhibits breast cancer stem cells in vitro and in vivo. Cancer Prev. Res. 2013, 6, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Warin, R.; Xiao, D.; Arlotti, J.A.; Bommareddy, A.; Singh, S.V. Inhibition of human breast cancer xenograft growth by cruciferous vegetable constituent benzyl isothiocyanate. Mol. Carcinog. 2010, 49, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sehrawat, A.; Singh, S.V. Notch2 activation by benzyl isothiocyanate impedes its inhibitory effect on breast cancer cell migration. Breast Cancer Res. Treat. 2012, 134, 1067–1079. [Google Scholar] [CrossRef]
- Antony, M.L.; Kim, S.-H.; Singh, S.V. Critical role of p53 upregulated modulator of apoptosis in benzyl isothiocyanate-induced apoptotic cell death. PLoS ONE 2012, 7, e32267. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Meng, Y.; Huang, L. Benzyl isothiocyanate inhibits breast cancer cell tumorigenesis via repression of the FoxH1-Mediated Wnt/β-catenin pathway. Int. J. Clin. Exp. Med. 2015, 8, 17601. [Google Scholar]
- Xie, B.; Nagalingam, A.; Kuppusamy, P.; Muniraj, N.; Langford, P.; Győrffy, B.; Saxena, N.K.; Sharma, D. Benzyl Isothiocyanate potentiates p53 signaling and antitumor effects against breast cancer through activation of p53-LKB1 and p73-LKB1 axes. Sci. Rep. 2017, 7, 40070. [Google Scholar] [CrossRef]
- Miyoshi, N.; Watanabe, E.; Osawa, T.; Okuhira, M.; Murata, Y.; Ohshima, H.; Nakamura, Y. ATP depletion alters the mode of cell death induced by benzyl isothiocyanate. Biochim. Biophys. Acta 2008, 1782, 566–573. [Google Scholar] [CrossRef]
- Yu, T.T.; Chang, M.Y.; Hsieh, Y.J.; Chang, C.J. Suppression of multiple processes relevant to cancer progression by benzyl isothiocyanate may result from the inhibition of Aurora A kinase activity. Food Funct. 2020, 11, 9010–9019. [Google Scholar] [CrossRef]
- Sarkar, R.; Mukherjee, S.; Biswas, J.; Roy, M. Phenethyl isothiocyanate, by virtue of its antioxidant activity, inhibits invasiveness and metastatic potential of breast cancer cells: HIF-1alpha as a putative target. Free Radic. Res. 2016, 50, 84–100. [Google Scholar] [CrossRef]
- Sarkars, R.; Mukherjee, S.; Roy, M. Targeting heat shock proteins by phenethyl isothiocyanate results in cell-cycle arrest and apoptosis of human breast cancer cells. Nutr. Cancer 2013, 65, 480–493. [Google Scholar] [CrossRef]
- Kang, L.; Wang, Z.Y. Breast cancer cell growth inhibition by phenethyl isothiocyanate is associated with down-regulation of oestrogen receptor-alpha36. J. Cell. Mol. Med. 2010, 14, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Y.T.; Moon, J.Y.; Ediriweera, M.K.; Cho, S.K. Phenethyl Isothiocyanate Suppresses Stemness in the Chemo- and Radio-Resistant Triple-Negative Breast Cancer Cell Line MDA-MB-231/IR Via Downregulation of Metadherin. Cancers 2020, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Singh, S.V. Bim contributes to phenethyl isothiocyanate-induced apoptosis in breast cancer cells. Mol. Carcinog. 2012, 51, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Cho, M.K. Phenethyl isothiocyanate induced apoptosis via down regulation of Bcl-2/XIAP and triggering of the mitochondrial pathway in MCF-7 cells. Arch. Pharm. Res. 2008, 31, 1604–1612. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, W.; Hao, M. Phenethyl isothiocyanate reduces breast cancer stem cell-like properties by epigenetic reactivation of CDH1. Oncol. Rep. 2021, 45, 337–348. [Google Scholar] [CrossRef]
- Gupta, P.; Wright, S.E.; Srivastava, S.K. PEITC treatment suppresses myeloid derived tumor suppressor cells to inhibit breast tumor growth. Oncoimmunology 2015, 4, e981449. [Google Scholar] [CrossRef]
- Huong, L.D.; Shim, J.H.; Choi, K.H.; Shin, J.A.; Choi, E.S.; Kim, H.S.; Lee, S.J.; Kim, S.J.; Cho, N.P.; Cho, S.D. Effect of beta-phenylethyl isothiocyanate from cruciferous vegetables on growth inhibition and apoptosis of cervical cancer cells through the induction of death receptors 4 and 5. J. Agric. Food Chem. 2011, 59, 8124–8131. [Google Scholar] [CrossRef]
- Zhang, L.; Hao, Q.; Bao, L.; Liu, W.; Fu, X.; Chen, Y.; Wu, H. Phenethyl isothiocyanate suppresses cervical carcinoma metastasis potential and its molecular mechanism. Mol. Med. Rep. 2014, 10, 2675–2680. [Google Scholar] [CrossRef]
- Upadhyaya, B.; Liu, Y.; Dey, M. Phenethyl Isothiocyanate Exposure Promotes Oxidative Stress and Suppresses Sp1 Transcription Factor in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 1027. [Google Scholar] [CrossRef]
- Shao, W.Y.; Yang, Y.L.; Yan, H.; Huang, Q.; Liu, K.J.; Zhang, S. Phenethyl isothiocyanate suppresses the metastasis of ovarian cancer associated with the inhibition of CRM1-mediated nuclear export and mTOR-STAT3 pathway. Cancer Biol. Ther. 2017, 18, 26–35. [Google Scholar] [CrossRef]
- Koschorke, A.; Faraci, S.; Giani, D.; Chiodoni, C.; Iorio, E.; Canese, R.; Colombo, M.P.; Lamolinara, A.; Iezzi, M.; Ladomery, M.; et al. Phenethyl isothiocyanate hampers growth and progression of HER2-positive breast and ovarian carcinoma by targeting their stem cell compartment. Cell. Oncol. 2019, 42, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.; Kandala, P.K.; Gupta, P.; Srivastava, S.K. Inhibition of EGFR-AKT axis results in the suppression of ovarian tumors in vitro and in preclinical mouse model. PLoS ONE 2012, 7, e43577. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Miskimins, W.K. Metformin and phenethyl isothiocyanate combined treatment in vitro is cytotoxic to ovarian cancer cultures. J. Ovarian Res. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Uddin, M.H.; Jo, U.; Kim, B.; Song, J.; Suh, D.H.; Kim, H.S.; Song, Y.S. ROS Accumulation by PEITC Selectively Kills Ovarian Cancer Cells via UPR-Mediated Apoptosis. Front. Oncol. 2015, 5, 167. [Google Scholar] [CrossRef]
- Bianco, R.; Shin, I.; Ritter, C.A.; Yakes, F.M.; Basso, A.; Rosen, N.; Tsurutani, J.; Dennis, P.A.; Mills, G.B.; Arteaga, C.L. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 2003, 22, 2812–2822. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Sestili, P.; Paolillo, M.; Lenzi, M.; Colombo, E.; Vallorani, L.; Casadei, L.; Martinelli, C.; Fimognari, C. Sulforaphane induces DNA single strand breaks in cultured human cells. Mutat. Res. Mol. Mech. Mutagen. 2010, 689, 65–73. [Google Scholar] [CrossRef]
- Hać, A.; Brokowska, J.; Rintz, E.; Bartkowski, M.; Węgrzyn, G.; Herman-Antosiewicz, A. Mechanism of selective anticancer activity of isothiocyanates relies on differences in DNA damage repair between cancer and healthy cells. Eur. J. Nutr. 2020, 59, 1421–1432. [Google Scholar] [CrossRef]
- Azarenko, O.; Okouneva, T.; Singletary, K.W.; Jordan, M.A.; Wilson, L. Suppression of microtubule dynamic instability and turnover in MCF7 breast cancer cells by sulforaphane. Carcinogenesis 2008, 29, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Kanematsu, S.; Yoshizawa, K.; Uehara, N.; Miki, H.; Sasaki, T.; Kuro, M.; Lai, Y.C.; Kimura, A.; Yuri, T.; Tsubura, A. Sulforaphane inhibits the growth of KPL-1 human breast cancer cells in vitro and suppresses the growth and metastasis of orthotopically transplanted KPL-1 cells in female athymic mice. Oncol. Rep. 2011, 26, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/beta-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Sadrieh, L.; Priyani, A.; Ahmed, M.; Hassan, A.H.; Hussain, A.J.C.E. Anti-carcinogenic effects of sulforaphane in association with its apoptosis-inducing and anti-inflammatory properties in human cervical cancer cells. Cancer Epidemiol. 2011, 35, 272–278. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef]
- Chuang, L.T.; Moqattash, S.T.; Gretz, H.F.; Nezhat, F.; Rahaman, J.; Chiao, J.W. Sulforaphane induces growth arrest and apoptosis in human ovarian cancer cells. Acta Obstet. Gynecol. Scand. 2007, 86, 1263–1268. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sung, B. NF-kappaB in cancer: A matter of life and death. Cancer Discov. 2011, 1, 469–471. [Google Scholar] [CrossRef]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef]
- Lamy, E.; Oey, D.; Eissmann, F.; Herz, C.; Munstedt, K.; Tinneberg, H.R.; Mersch-Sundermann, V. Erucin and benzyl isothiocyanate suppress growth of late stage primary human ovarian carcinoma cells and telomerase activity in vitro. Phytother. Res. 2013, 27, 1036–1041. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69 (Suppl. S3), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Syed Alwi, S.S.; Cavell, B.E.; Donlevy, A.; Packham, G. Differential induction of apoptosis in human breast cancer cell lines by phenethyl isothiocyanate, a glutathione depleting agent. Cell Stress Chaperones 2012, 17, 529–538. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.J.; Brazeau, D.A.; Morris, M.E. Dietary phenethyl isothiocyanate alters gene expression in human breast cancer cells. Evid.-Based Complement. Altern. Med. 2011, 2011, 462525. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Syed, V. TGF-beta Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Shoaib, S.; Tufail, S.; Sherwani, M.A.; Yusuf, N.; Islam, N. Phenethyl Isothiocyanate Induces Apoptosis Through ROS Generation and Caspase-3 Activation in Cervical Cancer Cells. Front. Pharmacol. 2021, 12, 673103. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Kim, M.C.; Chen, J.; Song, J.; Ko, H.W.; Lee, H.J. Sulforaphene interferes with human breast cancer cell migration and invasion through inhibition of hedgehog signaling. J. Agric. Food Chem. 2016, 64, 5515–5524. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-C.; Chang, M.-Y.; Wang, M.-J.; Yu, F.-S.; Liu, H.-C.; Harnod, T.; Hung, C.-H.; Lee, H.-T.; Chung, J.-G. PEITC inhibits human brain glioblastoma GBM 8401 cell migration and invasion through the inhibition of uPA, Rho A, and Ras with inhibition of MMP-2,-7 and-9 gene expression. Oncol. Rep. 2015, 34, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-J.; Cho, H.-J.; Chung, F.-L.; Wang, X.; Hoe, H.-S.; Park, K.-K.; Kim, C.-H.; Chang, H.-W.; Lee, S.-R.; Chang, Y.-C. Isothiocyanates suppress the invasion and metastasis of tumors by targeting FAK/MMP-9 activity. Oncotarget 2017, 8, 63949. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, Q. The protective effect of peroxiredoxin II on oxidative stress induced apoptosis in pancreatic β-cells. Cell Biosci. 2012, 2, 22. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Curtin, J.F.; Donovan, M.; Cotter, T.G. Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 2002, 265, 49–72. [Google Scholar] [CrossRef]
- Franco, R.; Sánchez-Olea, R.; Reyes-Reyes, E.M.; Panayiotidis, M.I. Environmental toxicity, oxidative stress and apoptosis: Menage a trois. Mutat. Res. 2009, 674, 3–22. [Google Scholar] [CrossRef]
- Kalkunte, S.; Swamy, N.; Dizon, D.S.; Brard, L. Benzyl isothiocyanate (BITC) induces apoptosis in ovarian cancer cells in vitro. J. Exp. Ther. Oncol. 2006, 5, 287–300. [Google Scholar]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Notte, A.; Leclere, L.; Michiels, C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 2011, 82, 427–434. [Google Scholar] [CrossRef]
- Liu, F.; Liu, D.; Yang, Y.; Zhao, S. Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol. Lett. 2013, 5, 1261–1265. [Google Scholar] [CrossRef]
- Xue, C.; Pasolli, H.A.; Piscopo, I.; Gros, D.J.; Liu, C.; Chen, Y.; Chiao, J.W. Mitochondrial structure alteration in human prostate cancer cells upon initial interaction with a chemopreventive agent phenethyl isothiocyanate. J. Cancer Cell. Int. 2014, 14, 30. [Google Scholar] [CrossRef]
- Bommareddy, A.; Hahm, E.-R.; Xiao, D.; Powolny, A.A.; Fisher, A.L.; Jiang, Y.; Singh, S.V. Atg5 Regulates Phenethyl Isothiocyanate–Induced Autophagic and Apoptotic Cell Death in Human Prostate Cancer Cells. J. Cancer Res. 2009, 69, 3704–3712. [Google Scholar] [CrossRef]
- Powolny, A.A.; Bommareddy, A.; Hahm, E.-R.; Normolle, D.P.; Beumer, J.H.; Nelson, J.B.; Singh, S.V. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J. Natl. Cancer Inst. 2011, 103, 571–584. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Castro, N.P.; Rangel, M.C.; Merchant, A.S.; MacKinnon, G.; Cuttitta, F.; Salomon, D.S.; Kim, Y.S. Sulforaphane Suppresses the Growth of Triple-negative Breast Cancer Stem-like Cells In vitro and In vivo. Cancer Prev. Res. 2019, 12, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Simoes, B.M.; Santiago-Gomez, A.; Chiodo, C.; Moreira, T.; Conole, D.; Lovell, S.; Alferez, D.; Eyre, R.; Spence, K.; Sarmiento-Castro, A.; et al. Targeting STAT3 signaling using stabilised sulforaphane (SFX-01) inhibits endocrine resistant stem-like cells in ER-positive breast cancer. Oncogene 2020, 39, 4896–4908. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Singh, S.V. The role of polycomb group protein Bmi-1 and Notch4 in breast cancer stem cell inhibition by benzyl isothiocyanate. Breast Cancer Res. Treat. 2015, 149, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Singh, S.V. Role of Kruppel-like Factor 4-p21(CIP1) Axis in Breast Cancer Stem-like Cell Inhibition by Benzyl Isothiocyanate. Cancer Prev. Res. 2019, 12, 125–134. [Google Scholar] [CrossRef]
- Kaczynska, A.; Swierczynska, J.; Herman-Antosiewicz, A. Sensitization of HER2 Positive Breast Cancer Cells to Lapatinib Using Plants-Derived Isothiocyanates. Nutr. Cancer 2015, 67, 976–986. [Google Scholar] [CrossRef]
- Kaczynska, A.; Herman-Antosiewicz, A. Combination of lapatinib with isothiocyanates overcomes drug resistance and inhibits migration of HER2 positive breast cancer cells. Breast Cancer 2017, 24, 271–280. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef]
- Royston, K.J.; Udayakumar, N.; Lewis, K.; Tollefsbol, T.O. A Novel Combination of Withaferin A and Sulforaphane Inhibits Epigenetic Machinery, Cellular Viability and Induces Apoptosis of Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1092. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, H.J.; Moon, D.O. Sulforaphane sensitizes human breast cancer cells to paclitaxel-induced apoptosis by downregulating the NF-kappaB signaling pathway. Oncol Lett. 2017, 13, 4427–4432. [Google Scholar] [CrossRef]
- Milczarek, M.; Wiktorska, K.; Mielczarek, L.; Koronkiewicz, M.; Dabrowska, A.; Lubelska, K.; Matosiuk, D.; Chilmonczyk, Z. Autophagic cell death and premature senescence: New mechanism of 5-fluorouracil and sulforaphane synergistic anticancer effect in MDA-MB-231 triple negative breast cancer cell line. Food Chem. Toxicol. 2018, 111, 1–8. [Google Scholar] [CrossRef]
- Wang, X.F.; Wu, D.M.; Li, B.X.; Lu, Y.J.; Yang, B.F. Synergistic inhibitory effect of sulforaphane and 5-fluorouracil in high and low metastasis cell lines of salivary gland adenoid cystic carcinoma. Phytother. Res. 2009, 23, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Mohsin, J.; Prabhu, S.A.; Begum, S.; Nusri Qel, A.; Harish, G.; Javed, E.; Khan, M.A.; Sharma, C. Sulforaphane inhibits growth of human breast cancer cells and augments the therapeutic index of the chemotherapeutic drug, gemcitabine. Asian Pac. J. Cancer Prev. 2013, 14, 5855–5860. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Sharma, S.; Sharma, A.; Vora, J.; Shrivastava, N. Sulforaphane-cisplatin combination inhibits the stemness and metastatic potential of TNBCs via down regulation of sirtuins-mediated EMT signaling axis. Phytomedicine 2021, 84, 153492. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Priyani, A.; Sadrieh, L.; Brahmbhatt, K.; Ahmed, M.; Sharma, C. Concurrent sulforaphane and eugenol induces differential effects on human cervical cancer cells. Integr. Cancer Ther. 2012, 11, 154–165. [Google Scholar] [CrossRef]
- Hunakova, L.; Gronesova, P.; Horvathova, E.; Chalupa, I.; Cholujova, D.; Duraj, J.; Sedlak, J. Modulation of cisplatin sensitivity in human ovarian carcinoma A2780 and SKOV3 cell lines by sulforaphane. Toxicol. Lett. 2014, 230, 479–486. [Google Scholar] [CrossRef]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Tollefsbol, T.O. Epigallocatechin gallate and sulforaphane combination treatment induce apoptosis in paclitaxel-resistant ovarian cancer cells through hTERT and Bcl-2 down-regulation. Exp. Cell Res. 2013, 319, 697–706. [Google Scholar] [CrossRef]
- Sharma, M.; Tollefsbol, T.O. Combinatorial epigenetic mechanisms of sulforaphane, genistein and sodium butyrate in breast cancer inhibition. Exp. Cell Res. 2022, 416, 113160. [Google Scholar] [CrossRef]
- Tian, M.; Tian, D.; Qiao, X.; Li, J.; Zhang, L. Modulation of Myb-induced NF-kB -STAT3 signaling and resulting cisplatin resistance in ovarian cancer by dietary factors. J. Cell Physiol. 2019, 234, 21126–21134. [Google Scholar] [CrossRef]
- Gong, T.T.; Liu, X.D.; Zhan, Z.P.; Wu, Q.J. Sulforaphane enhances the cisplatin sensitivity through regulating DNA repair and accumulation of intracellular cisplatin in ovarian cancer cells. Exp. Cell Res. 2020, 393, 112061. [Google Scholar] [CrossRef]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Tollefsbol, T.O. Enhancement of Cisplatin-Mediated Apoptosis in Ovarian Cancer Cells through Potentiating G2/M Arrest and p21 Upregulation by Combinatorial Epigallocatechin Gallate and Sulforaphane. J. Oncol. 2013, 2013, 872957. [Google Scholar] [CrossRef]
- Bose, C.; Awasthi, S.; Sharma, R.; Benes, H.; Hauer-Jensen, M.; Boerma, M.; Singh, S.P. Sulforaphane potentiates anticancer effects of doxorubicin and attenuates its cardiotoxicity in a breast cancer model. PLoS ONE 2018, 13, e0193918. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Govind, S.; Sajankila, S.P.; Mi, L.; Roy, R.; Chung, F.L. Phenethyl isothiocyanate sensitizes human cervical cancer cells to apoptosis induced by cisplatin. Mol. Nutr. Food Res. 2011, 55, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dey, S.; Bhattacharya, R.K.; Roy, M. Isothiocyanates sensitize the effect of chemotherapeutic drugs via modulation of protein kinase C and telomerase in cervical cancer cells. Mol. Cell Biochem. 2009, 330, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Ma, Y.; Chiao, J.W.; Liu, D. Phenethyl isothiocyanate and paclitaxel synergistically enhanced apoptosis and alpha-tubulin hyperacetylation in breast cancer cells. Exp. Hematol. Oncol. 2014, 3, 5. [Google Scholar] [CrossRef]
- Gupta, P.; Srivastava, S.K. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med. 2012, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cang, S.; Ma, Y.; Chiao, J.W. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer Cell. Int. 2013, 13, 10. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, M.; Sang, X.; Liu, P.; Gao, J.; Jiang, K.; Cheng, H. Phenethyl Isothiocyanate Enhances the Cytotoxic Effects of PARP Inhibitors in High-Grade Serous Ovarian Cancer Cells. Front. Oncol. 2021, 11, 812264. [Google Scholar] [CrossRef]
- Eisa, N.H.; ElSherbiny, N.M.; Shebl, A.M.; Eissa, L.A.; El-Shishtawy, M.M. Phenethyl isothiocyanate potentiates anti-tumour effect of doxorubicin through Akt-dependent pathway. Cell Biochem. Funct. 2015, 33, 541–551. [Google Scholar] [CrossRef]
- Paul, B.; Li, Y.; Tollefsbol, T.O. The Effects of Combinatorial Genistein and Sulforaphane in Breast Tumor Inhibition: Role in Epigenetic Regulation. Int. J. Mol. Sci. 2018, 19, 1754. [Google Scholar] [CrossRef]
- Warin, R.; Chambers, W.H.; Potter, D.M.; Singh, S.V. Prevention of mammary carcinogenesis in MMTV-neu mice by cruciferous vegetable constituent benzyl isothiocyanate. Cancer Res. 2009, 69, 9473–9480. [Google Scholar] [CrossRef]
- Sehrawat, A.; Croix, C.S.; Baty, C.J.; Watkins, S.; Tailor, D.; Singh, R.P.; Singh, S.V. Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion 2016, 30, 67–77. [Google Scholar] [CrossRef]
- Kim, E.J.; Hong, J.E.; Eom, S.J.; Lee, J.Y.; Park, J.H. Oral administration of benzyl-isothiocyanate inhibits solid tumor growth and lung metastasis of 4T1 murine mammary carcinoma cells in BALB/c mice. Breast Cancer Res. Treat. 2011, 130, 61–71. [Google Scholar] [CrossRef]
- Xie, B.; Zhao, L.; Guo, L.; Liu, H.; Fu, S.; Fan, W.; Lin, L.; Chen, J.; Wang, B.; Fan, L.; et al. Benzyl isothiocyanate suppresses development and metastasis of murine mammary carcinoma by regulating the Wnt/beta-catenin pathway. Mol. Med. Rep. 2019, 20, 1808–1818. [Google Scholar] [CrossRef]
- Kim, M.; Cho, H.J.; Kwon, G.T.; Kang, Y.H.; Kwon, S.H.; Her, S.; Park, T.; Kim, Y.; Kee, Y.; Park, J.H. Benzyl isothiocyanate suppresses high-fat diet-stimulated mammary tumor progression via the alteration of tumor microenvironments in obesity-resistant BALB/c mice. Mol. Carcinog. 2015, 54, 72–82. [Google Scholar] [CrossRef]
- Gupta, P.; Adkins, C.; Lockman, P.; Srivastava, S.K. Metastasis of Breast Tumor Cells to Brain Is Suppressed by Phenethyl Isothiocyanate in a Novel In Vivo Metastasis Model. PLoS ONE 2013, 8, e67278. [Google Scholar] [CrossRef]
- Wang, Z.; Tu, C.; Pratt, R.; Khoury, T.; Qu, J.; Fahey, J.W.; McCann, S.E.; Zhang, Y.; Wu, Y.; Hutson, A.D.; et al. A Presurgical-Window Intervention Trial of Isothiocyanate-Rich Broccoli Sprout Extract in Patients with Breast Cancer. Mol. Nutr. Food Res. 2022, 66, e2101094. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Behavioral Dietary Intervention for the Improvement of Bladder Cancer Survivorship. Available online: https://ClinicalTrials.gov/show/NCT04548193 (accessed on 21 March 2023).
- Clinicaltrials.gov. A Study of the Effects of PEITC on Oral Cells with Mutant p53. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01790204 (accessed on 22 March 2023).
- Clinicaltrials.gov. Chemoprevention of Prostate Cancer, HDAC Inhibition and DNA Methylation (PBroC). Available online: https://clinicaltrials.gov/ct2/show/NCT01265953 (accessed on 24 March 2023).
- Lamy, E.; Garcia-Kaufer, M.; Prinzhorn, J.; Mersch-Sundermann, V. Antigenotoxic action of isothiocyanate-containing mustard as determined by two cancer biomarkers in a human intervention trial. Eur. J. Cancer Prev. 2012, 21, 400–406. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Phenethyl Isothiocyanate in Preventing Lung Cancer in Smokers. Available online: https://clinicaltrials.gov/ct2/show/NCT00691132 (accessed on 25 March 2023).
- Clinicaltrials.gov. Phenethyl Isothiocyanate in Preventing Lung Cancer in People Who Smoke. Available online: https://clinicaltrials.gov/ct2/show/NCT00005883 (accessed on 27 March 2023).


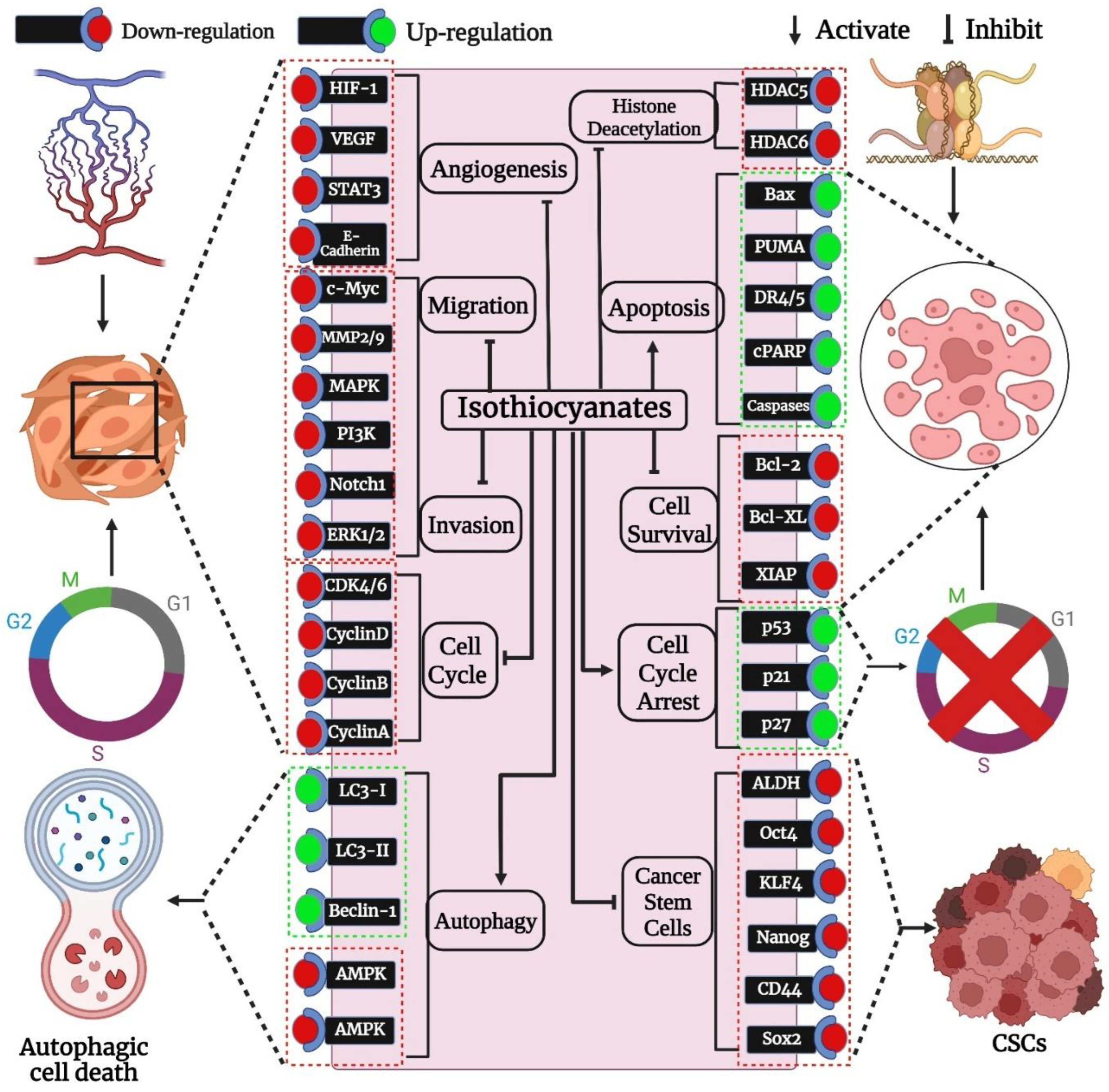{kind=link}
| Isothiocyanates | Structure | Sources of the Compound |
|---|---|---|
| Sulforaphane (SFN) |  | Broccoli, watercress, cauliflower, cabbage, kale, brussels sprouts, broccoli sprouts, etc. |
| Benzyl isothiocyanate (BITC) |  | Broccoli, watercress, cauliflower, cabbage, pink mustard, papaya seeds, pilu tree, etc. |
| Phenethyl isothiocyanate (PEITC) |  | Broccoli, watercress, radish, turnip, cauliflower, cabbage, etc. |
| Phytochemicals | Cancer Model Studied | Molecular Mechanism | Major Findings | References |
|---|---|---|---|---|
| Sulforaphane (SFN) | MCF-7, MDA-MB-231, SKBR-3 and MDA MB 468 | Decreased phosphorylation of Akt and S6K1 | Growth Inhibition | [70] |
| MCF-7, MDA-MB-231 | Suppression of hTERT and down-regulation of DNMT1 and DNMT3a | Proliferation Inhibition | [71] | |
| MCF-7 | Inhibition of estrogen receptor alpha protein and inhibition of progesterone receptor | Proliferation Inhibition | [72] | |
| MCF-7, MDA-MB-231 | Activation of Caspase-3, Bax, and p21, and down-regulation of Bcl-2, cyclin A, cyclin B1, and Cdc2 | Apoptosis, Cell Cycle Arrest Autophagy Inhibition | [73] | |
| MDA-MB-468, MCF-7 and T47D | Down-regulation of EGFR, HDAC, Bcl-2, caspase-3, and caspase-9 | Apoptosis | [74] | |
| MCF-7 and SUM159 | Down-regulation of Wnt/β-catenin | Apoptosis Induction, Cell Viability Inhibition, CSCs Inhibition | [75] | |
| MDA-MB-231, MCF-7 and SKBR-3 | Down-regulation of Akt and DNMT and activation of p21 and p27 | Apoptosis, Cell Cycle Arrest | [76] | |
| MDA-MB-468, MCF-7 and BT-474 | Inhibition of HDAC5 and LSD1 Axis | Growth Inhibition | [77] | |
| MCF-7 | Suppression of NF-κB signaling pathway and inhibition of TPA-induced MMP-9 expression | Proliferation Inhibition | [78] | |
| MDA-MB-231, BT549 and MDA-MB-468 | Down-regulation of HDAC6 and increased membrane translocation and acetylation modification of PTEN | Growth Inhibition and Modulation of Autophagy | [79] | |
| OVCAR3 and A2780 | Reduced Akt and NF-κB signaling; down-regulation of Bcl-2, Bcl-xL, c-Myc, and cyclin D1 | Apoptosis and Cell Cycle Arrest | [80] | |
| SKOV3 | Reduced Akt and PI3K signaling to control the expression of cyclin D1 and CDK4/6 | Anti-Proliferative and Cell Cycle Arrest | [39] | |
| PA-1 | Reduced cyclin B1 and Cdc2 expression | Cell Cycle Arrest | [81] | |
| MDAH2774 and SKOV3 | Enhanced activity of Rb, c-PARP, and increased Bak/Bcl-2 ratio | Anti-Proliferative, Apoptosis Induction, and Cell Cycle Arrest | [82] | |
| A2780 and A2780/CP | Reduced AP1 and HIF-1 expression | Anti-proliferative and Anti-Metastatic | [83] | |
| HeLa, Cx, and CxWJ | Down-regulated cyclin B1 and up-regulated GADD45β | Growth Inhibition and Cell Cycle Arrest | [84] | |
| HeLa | Down-regulated Bcl-2, IL-1β, and COX-2 | Growth Inhibition, Apoptosis, and Anti-Inflammatory | [84] | |
| HeLa | Targeted HDAC and DNMT3b | Apoptosis and Cell Cycle Arrest | [85] | |
| HeLa | Promoted Bax, caspase-3, and PARP cleavage while inhibited Bcl-2 and Bcl-xL | Apoptosis | [86] | |
| Benzyl isothiocyanate (BITC) | MCF-7 and MDA-MB-231 | Targeted JNK and p38 MAPK to generate excessive ROS activating Bax and Caspase-3 | Apoptosis and Growth Inhibition | [87] |
| MCF-7 and MDA-MB-231 | Down-regulation of Bcl-xL, Bcl-2, cyclin B1, and CDK1, and up-regulation of Bax and Bak expression | Cell Growth Inhibition, Apoptosis, and Cell Cycle Arrest | [88] | |
| MDA-MB-231 and in vivo | Down-regulation of vimentin, fibronectin, Snail, and c-Met | Inhibition of EMT | [89] | |
| SUM159, MDA-MB-231 and in vivo | Up-regulation of E-cadherin and repressed uPA along with vimentin expression inhibition | Anti-Metastatic and Inhibition of EMT | [90] | |
| MCF-7 | Promoted LC3 cleavage and reduced mTOR and p68 expression | Autophagy Cell Death and Growth Inhibition | [91] | |
| MCF-7 and in vivo | Inhibited expression of vimentin and N-cadherin and overexpressed E-cadherin | Inhibition of CSCs | [92] | |
| MDA-MB-231 and in vivo | Down-regulation of VEGF receptor-2 | Anti-metastatic and Anti-Angiogenic | [93] | |
| MCF-7, SUM159 and MDA-MB-231 | Activation of Notch2 signaling | Anti-Metastatic and Anti-Proliferative | [94] | |
| MCF7, MDA-MB-231 | Down-regulation of Bcl-xL and Bcl-2 and up-regulation of PUMA | Apoptosis and Growth Inhibition | [95] | |
| MDA-MB-231 and MCF-7 | Down-regulation of FOXH1 and Wnt/β-catenin | Growth and Invasion Inhibition | [96] | |
| MCF-7 | Activation of p53-LKB1 and p73-LKB1 axes | Growth Inhibition | [97] | |
| HeLa | Reduced ATP levels and cause DNA fragmentation | Growth Inhibition and Apoptosis | [98] | |
| HeLa | Inhibition of Aurora A and PLK1 expression | Cell Cycle Arrest | [99] | |
| Phenethyl Isothiocyanate (PEITC) | MCF-7 and MDA-MB-231 | Reduced expression of HIF-1α, VEGF, and MMP2/9 | Growth Inhibition | [100] |
| MCF-7 and MDA-MB-231 | Inhibition of HSPs (particularly HSP 90) and HSF1 Reduced expression of anti-apoptotic Bcl-2 protein, CDK1, and Cdc25C and increased expression of caspases, Bax, p21, and p53 | Apoptosis and Cell Cycle Arrest | [101] | |
| MCF-7, H3396, SKBR-3 and MDA-MB-231 | Down-regulation of estrogen receptor-α36 and abrogation of MAPK/ERK1/2 signaling | Growth Suppression | [102] | |
| MDA-MB-231/IR | Down-regulation of Metadherin, CD44, Slug, and β-catenin | Inhibition of CSCs | [103] | |
| BRI-JM04 MCF-7, and MDA-MB-231 | Up-regulation of Bak, PUMA, and Bim (long and short forms of Bim), increased S65 phosphorylation of BimEL (extra-long form), and down-regulation of Bcl-xL and Bcl-2 | Apoptosis and Growth Inhibition | [104] | |
| MCF-7 | Down-regulation of Bcl-2/XIAP, up-regulation of procaspase-7/-9, PARP cleavage | Apoptosis and Cell Survival Inhibition | [105] | |
| MCF-7 and MDA-MB-231 | Epigenetic reactivation of CDH1, down-regulation of Wnt/β-catenin signaling, and Inhibition of HDAC and DNMT expression | Inhibition of CSCs | [106] | |
| In vivo (Mice having MDA-MB-231 xenografts on MDSCs) | Inhibition of myeloid-derived suppressor cells (MDSCs) | Anti-Tumor Activity | [107] | |
| HEp-2 and KB | Activation of DR4 and DR5 through inactivation of ERK and MEK | Growth Inhibition and Apoptosis | [108] | |
| HeLa | Activation of TGF-β/Smad2 signaling pathway, reduced the expression of CDK1, MMP-2/9, CD44, and ICAM-1, increased the production of TGF-β, IL-6, and IL-8, and increased the phosphorylation of Smad2 | Anti-Metastasis and Cell Cycle Arrest | [109] | |
| HeLa | Inhibition of Sp1 transcription factor and downstream multidrug resistance protein (P-glycoprotein) | Inhibition of CSCs | [110] | |
| SKOV-3, HO8910 and in vivo | Suppression of MMPs and mTOR-STAT3 signaling | Anti-proliferative and Anti-Metastatic | [111] | |
| HER2-positive BT474, SKBR3, HCC1954, SKOV3 and in vivo | Reduction in Notch1 and HER2 expression | Inhibition of CSCs, and Growth Inhibition | [112] | |
| SKOV-3, OVCAR-3, TOV-21G and in vivo | Inhibition of EGFR and Akt | Growth Inhibition and Apoptosis | [113] | |
| SKOV-3, OVCAR-3 and CAOV-3 | Induced excessive ROS production | Apoptosis | [114] | |
| SKOV-3 and PA-1 | Up-regulation of the key regulator of UPR-mediated apoptosis, CHOP/GADD153, and endoplasmic reticulum resident chaperone BiP/GRP78 along with activation of two major sensors of the UPR (PERK and ATF-6 in PA-1; PERK and IRE1α in SKOV-3) | Apoptosis | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoaib, S.; Khan, F.B.; Alsharif, M.A.; Malik, M.S.; Ahmed, S.A.; Jamous, Y.F.; Uddin, S.; Tan, C.S.; Ardianto, C.; Tufail, S.; et al. Reviewing the Prospective Pharmacological Potential of Isothiocyanates in Fight against Female-Specific Cancers. Cancers 2023, 15, 2390. https://doi.org/10.3390/cancers15082390
Shoaib S, Khan FB, Alsharif MA, Malik MS, Ahmed SA, Jamous YF, Uddin S, Tan CS, Ardianto C, Tufail S, et al. Reviewing the Prospective Pharmacological Potential of Isothiocyanates in Fight against Female-Specific Cancers. Cancers. 2023; 15(8):2390. https://doi.org/10.3390/cancers15082390
Chicago/Turabian StyleShoaib, Shoaib, Farheen Badrealam Khan, Meshari A. Alsharif, M. Shaheer Malik, Saleh A. Ahmed, Yahya F. Jamous, Shahab Uddin, Ching Siang Tan, Chrismawan Ardianto, Saba Tufail, and et al. 2023. "Reviewing the Prospective Pharmacological Potential of Isothiocyanates in Fight against Female-Specific Cancers" Cancers 15, no. 8: 2390. https://doi.org/10.3390/cancers15082390
APA StyleShoaib, S., Khan, F. B., Alsharif, M. A., Malik, M. S., Ahmed, S. A., Jamous, Y. F., Uddin, S., Tan, C. S., Ardianto, C., Tufail, S., Ming, L. C., Yusuf, N., & Islam, N. (2023). Reviewing the Prospective Pharmacological Potential of Isothiocyanates in Fight against Female-Specific Cancers. Cancers, 15(8), 2390. https://doi.org/10.3390/cancers15082390