
PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma
 , , and
, , and 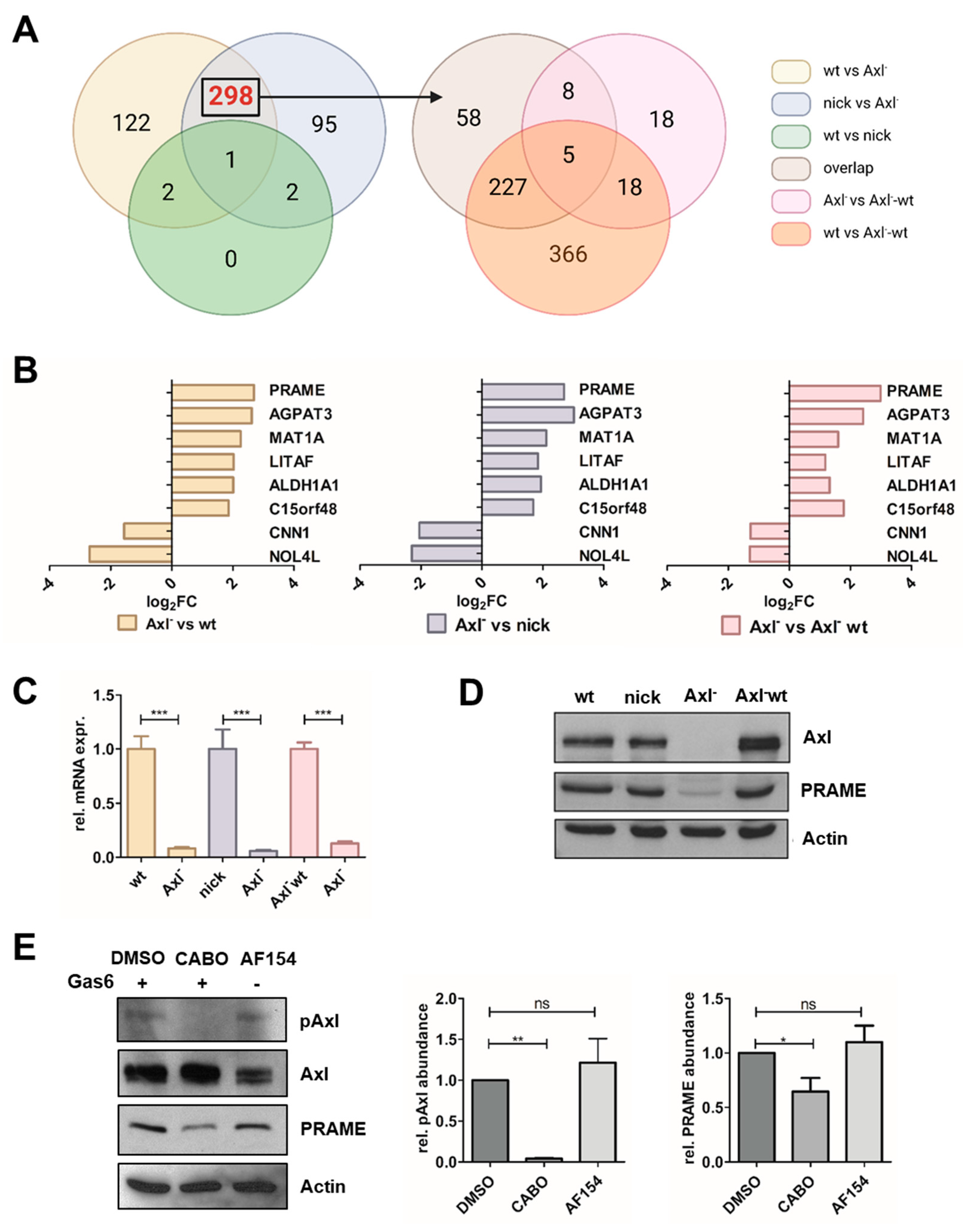{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Knockdown by RNA Interference
2.3. 2D Wound Healing Assay
2.4. 3D Invasion Assay
2.5. Clonogenic Survival Assay
2.6. Proliferation Kinetics
2.7. Transcriptome Profiling by RNA-seq
2.8. Reverse Transcription, Quantitative Polymerase Chain Reaction (RT-qPCR)
2.9. Western Blotting
2.10. Immunoprecipitation (IP)
2.11. Mass Spectrometry Analyses of IPs
2.12. Correlation Analysis
2.13. In Silico Analysis
2.14. Immunohistochemistry of Primary HCC Patient Samples
2.15. Statistical Analysis
3. Results
3.1. Identification of Gas6/Axl-Dependent Targets in HCC Cells
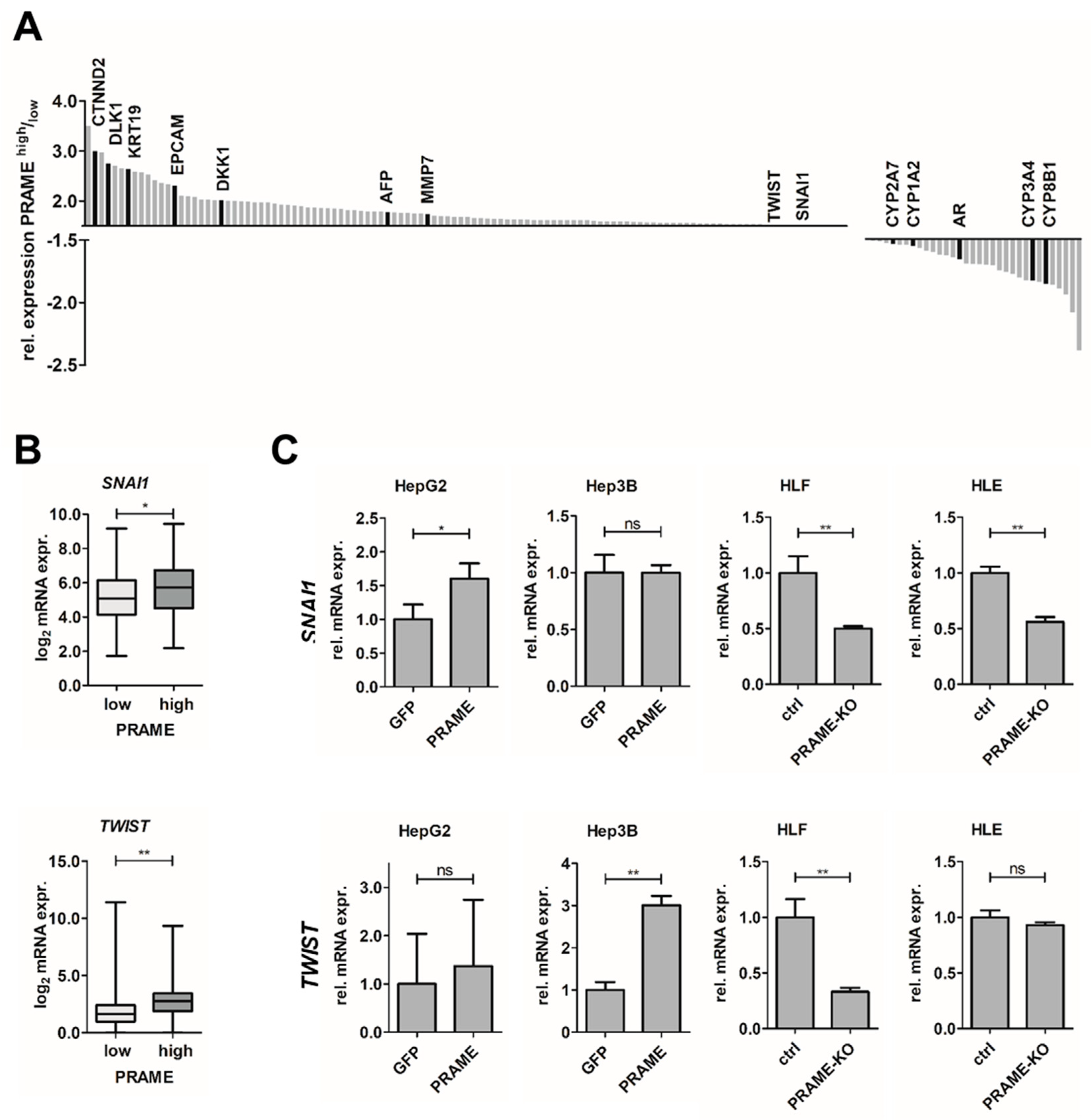
3.2. Expression of PRAME Correlates with Dedifferentiation of HCC Cells and EMT
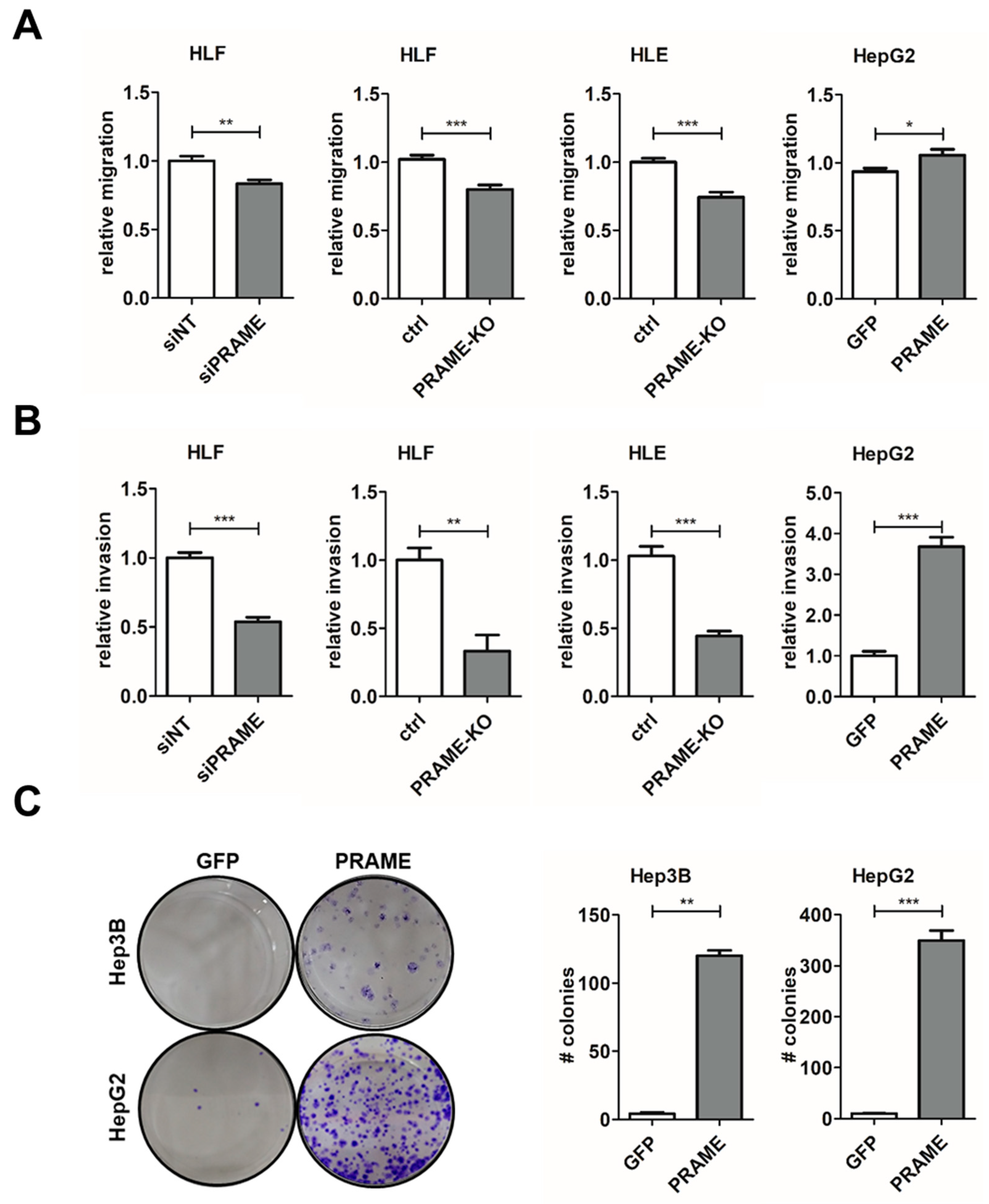
3.3. PRAME Augments 2D Cell Migration and 3D Invasion
3.4. Axl-Induced MAPK Signaling Modulates PRAME Expression
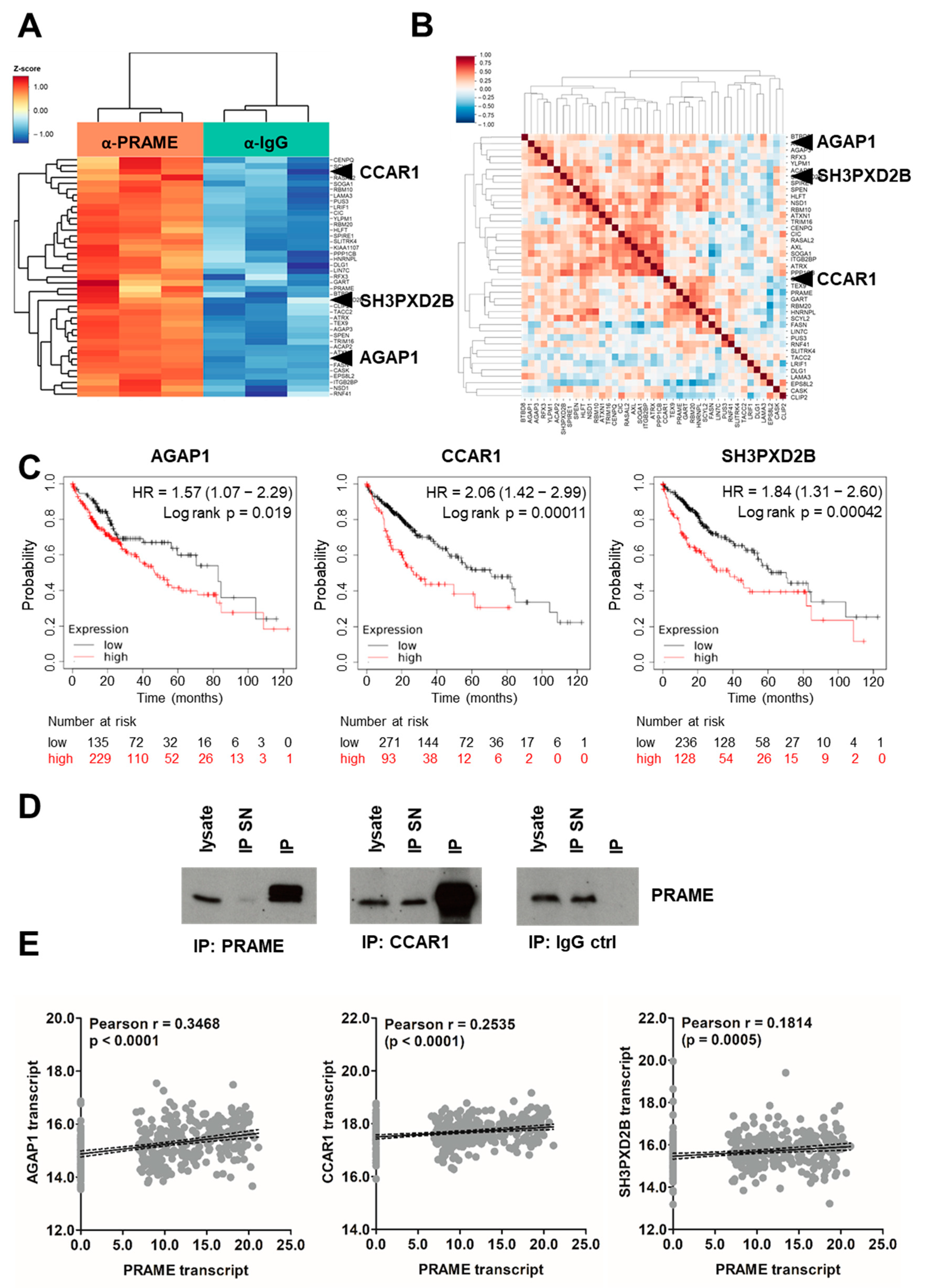
3.5. Identification of PRAME Binding Partners
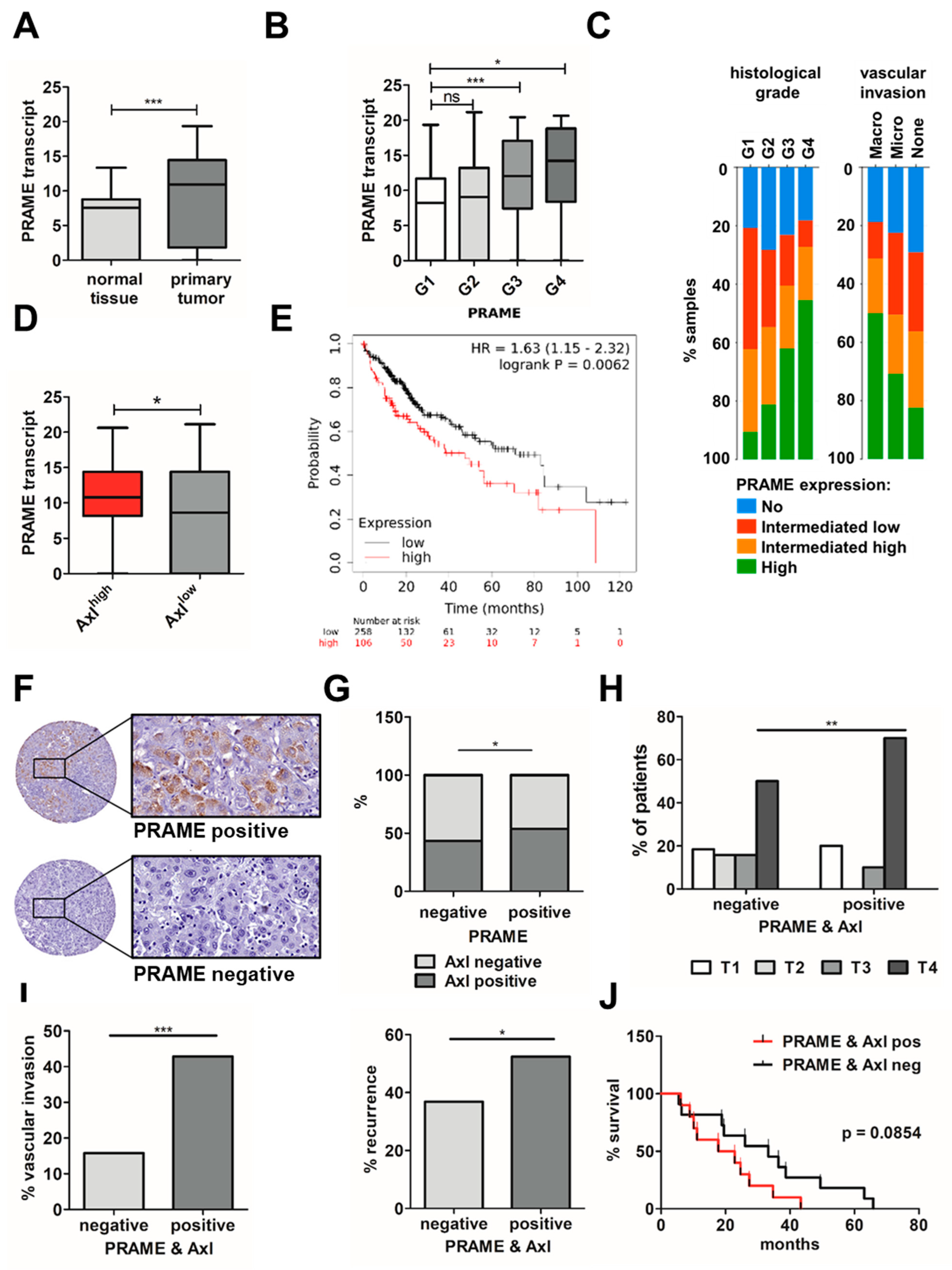
3.6. Expression of PRAME and Axl in HCC Patients Correlates with Advanced Stage, Vascular Invasion and Poor Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef]
- Llovet, J.M. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; Von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, V.; Breitenecker, K.; Djerlek, L.; Ortmayr, G.; Mikulits, W. Intrinsic and Extrinsic Control of Hepatocellular Carcinoma by TAM Receptors. Cancers 2021, 13, 5448. [Google Scholar] [CrossRef]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef]
- Golkowski, M.; Lau, H.-T.; Chan, M.; Kenerson, H.; Vidadala, V.N.; Shoemaker, A.; Maly, D.J.; Yeung, R.S.; Gujral, T.S.; Ong, S.-E. Pharmacoproteomics Identifies Kinase Pathways that Drive the Epithelial-Mesenchymal Transition and Drug Resistance in Hepatocellular Carcinoma. Cell Syst. 2020, 11, 196–207.e7. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Abu-Thuraia, A.; Goyette, M.-A.; Boulais, J.; Delliaux, C.; Apcher, C.; Schott, C.; Chidiac, R.; Bagci, H.; Thibault, M.-P.; Davidson, D.; et al. AXL confers cell migration and invasion by hijacking a PEAK1-regulated focal adhesion protein network. Nat. Commun. 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Pietrobono, S.; Anichini, G.; Sala, C.; Manetti, F.; Almada, L.L.; Pepe, S.; Carr, R.M.; Paradise, B.D.; Sarkaria, J.N.; Davila, J.I.; et al. ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Ikeda, H.; Lethé, B.; Lehmann, F.; Van Baren, N.; Baurain, J.-F.; De Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Epping, M.T.; Wang, L.; Edel, M.J.; Carlée, L.; Hernandez, M.; Bernards, R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 2005, 122, 835–847. [Google Scholar] [CrossRef]
- Amir, A.L.; van der Steen, D.M.; van Loenen, M.M.; Hagedoorn, R.S.; de Boer, R.; Kester, M.D.; de Ru, A.H.; Lugthart, G.-J.; van Kooten, C.; Hiemstra, P.S.; et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin. Cancer Res. 2011, 17, 5615–5625. [Google Scholar] [CrossRef]
- Kirkey, D.C.; Loeb, A.M.; Castro, S.; McKay, C.N.; Perkins, L.; Pardo, L.; Leonti, A.R.; Tang, T.T.; Loken, M.R.; Brodersen, L.E.; et al. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2023, 7, 1178–1189. [Google Scholar] [CrossRef]
- Epping, M.T.; Bernards, R. A causal role for the human tumor antigen preferentially expressed antigen of melanoma in cancer. Cancer Res 2006, 66, 10639–10642. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Sun, X.; Liu, Z.; He, Y. A novel era of cancer/testis antigen in cancer immunotherapy. Int. Immunopharmacol. 2021, 98, 107889. [Google Scholar] [CrossRef]
- Liu, K.; Cheng, L.; Zhu, K.; Wang, J.; Shu, Q. The cancer/testis antigen HORMAD1 mediates epithelial-mesenchymal transition to promote tumor growth and metastasis by activating the Wnt/beta-catenin signaling pathway in lung cancer. Cell Death Discov. 2022, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Qiao, Y.; Meng, M.; Zhou, Q. Cancer/Testis Antigens as Biomarker and Target for the Diagnosis, Prognosis, and Therapy of Lung Cancer. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Cai, L.; Liang, Y.; Xu, J.; Liu, Y.; Zhou, L.; Ding, C.; Zhang, Y.; Zhao, H.; et al. Tumor-associated antigen Prame targets tumor suppressor p14/ARF for degradation as the receptor protein of CRL2(Prame) complex. Cell Death Differ. 2021, 28, 1926–1940. [Google Scholar] [CrossRef] [PubMed]
- Al-Khadairi, G.; Naik, A.; Thomas, R.; Al-Sulaiti, B.; Rizly, S.; Decock, J. PRAME promotes epithelial-to-mesenchymal transition in triple negative breast cancer. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Cao, H.; Yang, J.-W.; Meng, W.-X.; Yang, C.; Wang, J.-T.; Yu, M.-M.; Wang, B.-S. HDAC5-mediated PRAME regulates the proliferation, migration, invasion, and EMT of laryngeal squamous cell carcinoma via the PI3K/AKT/mTOR signaling pathway. Open Med. 2023, 18. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Zweemer, A.J.; Lauffenburger, D.A. The AXL Receptor Is a Sensor of Ligand Spatial Heterogeneity. Cell Syst. 2015, 1, 25–36. [Google Scholar] [CrossRef]
- Gould, W.R.; Baxi, S.M.; Schroeder, R.; Peng, Y.W.; Leadley, R.J.; Peterson, J.T.; Perrin, L.A. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J. Thromb. Haemost. 2005, 3, 733–741. [Google Scholar] [CrossRef]
- Scharf, I.; Bierbaumer, L.; Huber, H.; Wittmann, P.; Haider, C.; Pirker, C.; Berger, W.; Mikulits, W. Dynamics of CRISPR/Cas9-mediated genomic editing of the AXL locus in hepatocellular carcinoma cells. Oncol. Lett. 2018, 15, 2441–2450. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-beta and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef]
- Osswald, A.; Hedrich, V.; Sommergruber, W. 3D-3 Tumor Models in Drug Discovery for Analysis of Immune Cell Infiltration. Methods Mol. Biol. 2019, 1953, 151–162. [Google Scholar] [CrossRef]
- Berens, E.B.; Holy, J.M.; Riegel, A.T.; Wellstein, A. A Cancer Cell Spheroid Assay to Assess Invasion in a 3D Setting. J. Vis. Exp. 2015, e53409. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Oyama, K.; Kanki, K.; Shimizu, H.; Kono, Y.; Azumi, J.; Toriguchi, K.; Hatano, E.; Shiota, G. Impact of Preferentially Expressed Antigen of Melanoma on the Prognosis of Hepatocellular Carcinoma. Gastrointest. Tumors 2017, 3, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Jung, E.; Alfonso, J.; Monyer, H.; Wick, W.; Winkler, F. Neuronal signatures in cancer. Int. J. Cancer 2020, 147, 3281–3291. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, T.; Zhang, S.; Guo, K.; Liu, Y. Oct4 induces EMT through LEF1/beta-catenin dependent WNT signaling pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tsai, Y.; Huang, Y.; Liang, Y.; Sun, Y.; Su, C.; Chau, G.; Yeh, Y.; Chang, Y.; Hu, J.; et al. Lymphoid Enhancer Factor 1 Contributes to Hepatocellular Carcinoma Progression Through Transcriptional Regulation of Epithelial-Mesenchymal Transition Regulators and Stemness Genes. Hepatol. Commun. 2018, 2, 1392–1407. [Google Scholar] [CrossRef]
- Naik, A.; Thomas, R.; Al-Khadairi, G.; Bacha, R.; Hendrickx, W.; Decock, J. Cancer testis antigen PRAME: An anti-cancer target with immunomodulatory potential. J. Cell. Mol. Med. 2021, 25, 10376–10388. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.-A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020, 183, 377–394.e21. [Google Scholar] [CrossRef]
- Ma, W.-L.; Hsu, C.-L.; Yeh, C.-C.; Wu, M.-H.; Huang, C.-K.; Jeng, L.-B.; Hung, Y.-C.; Lin, T.-Y.; Yeh, S.; Chang, C. Hepatic androgen receptor suppresses hepatocellular carcinoma metastasis through modulation of cell migration and anoikis. Hepatology 2012, 56, 176–185. [Google Scholar] [CrossRef]
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011, 475, 386–389. [Google Scholar] [CrossRef]
- Alder, O.; Cullum, R.; Lee, S.; Kan, A.C.; Wei, W.; Yi, Y.; Garside, V.C.; Bilenky, M.; Griffith, M.; Morrissy, A.S.; et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell Rep. 2014, 9, 261–271. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, D.D.; Mello, B.P.; Pereira, W.O.; Amarante-Mendes, G.P. PRAME/EZH2-mediated regulation of TRAIL: A new target for cancer therapy. Curr. Mol. Med. 2013, 13, 296–304. [Google Scholar] [CrossRef]
- Costessi, A.; Mahrour, N.; Tijchon, E.; Stunnenberg, R.; A Stoel, M.; Jansen, P.W.; Sela, D.; Martin-Brown, S.; Washburn, M.; Florens, L.; et al. The tumour antigen PRAME is a subunit of a Cul2 ubiquitin ligase and associates with active NFY promoters. EMBO J. 2011, 30, 3786–3798. [Google Scholar] [CrossRef] [PubMed]
- Wadelin, F.R.; Fulton, J.; Collins, H.M.; Tertipis, N.; Bottley, A.; Spriggs, K.A.; Falcone, F.H.; Heery, D.M. PRAME is a golgi-targeted protein that associates with the Elongin BC complex and is upregulated by interferon-gamma and bacterial PAMPs. PLoS ONE 2013, 8, e58052. [Google Scholar] [CrossRef]
- Wang, S.; Xia, W.; Qiu, M.; Wang, X.; Jiang, F.; Yin, R.; Xu, L. Atlas on substrate recognition subunits of CRL2 E3 ligases. Oncotarget 2016, 7, 46707–46716. [Google Scholar] [CrossRef]
- Costessi, A.; Mahrour, N.; Sharma, V.; Stunnenberg, R.; Stoel, M.A.; Tijchon, E.; Conaway, J.W.; Conaway, R.C.; Stunnenberg, H.G. The Human EKC/KEOPS Complex Is Recruited to Cullin2 Ubiquitin Ligases by the Human Tumour Antigen PRAME. PLOS ONE 2012, 7, e42822. [Google Scholar] [CrossRef] [PubMed]
- Baba, H.; Kanda, M.; Sawaki, K.; Umeda, S.; Miwa, T.; Shimizu, D.; Tanaka, C.; Kobayashi, D.; Fujiwara, M.; Kodera, Y.; et al. PRAME as a Potential Biomarker for Liver Metastasis of Gastric Cancer. Ann. Surg. Oncol. 2019, 27, 2071–2080. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, J.; Yin, J.; Lü, B.; Yang, Q.; Wan, Y.; Jia, C. Downregulation of PRAME Suppresses Proliferation and Promotes Apoptosis in Hepatocellular Carcinoma Through the Activation of P53 Mediated Pathway. Cell. Physiol. Biochem. 2018, 45, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef]
- Chang, T.-S.; Wei, K.-L.; Lu, C.-K.; Chen, Y.-H.; Cheng, Y.-T.; Tung, S.-Y.; Wu, C.-S.; Chiang, M.-K. Inhibition of CCAR1, a Coactivator of beta-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells. Int. J. Mol. Sci. 2017, 18, 460. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Luo, G.-J.; Ma, X.-K.; Lu, X.-Y.; Lin, X.-M.; Yang, S.; Zhao, Q.; Wu, T.; Li, Z.X.; et al. Circular RNAs negatively regulate cancer stem cells by physically binding FMRP against CCAR1 complex in hepatocellular carcinoma. Theranostics 2019, 9, 3526–3540. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, L.; Lin, Z.; Yu, D.; Jin, M.; Zhou, P.; Ren, J.; Cheng, J.; Yang, K.; Wu, G.; et al. Targeting DCLK1 overcomes 5-fluorouracil resistance in colorectal cancer through inhibiting CCAR1/beta-catenin pathway-mediated cancer stemness. Clin. Transl. Med. 2022, 12, e743. [Google Scholar] [PubMed]
- Iizuka, S.; Abdullah, C.; Buschman, M.D.; Diaz, B.; Courtneidge, S.A. The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma. Oncotarget 2016, 7, 78473–78486. [Google Scholar] [CrossRef] [PubMed]
- Quintanilha, J.C.F.; Wang, J.; Sibley, A.B.; Xu, W.; Espin-Garcia, O.; Jiang, C.; Etheridge, A.S.; Ratain, M.J.; Lenz, H.; Bertagnolli, M.; et al. Genome-wide association studies of survival in 1520 cancer patients treated with bevacizumab-containing regimens. Int. J. Cancer 2022, 150, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-S.; Lu, C.; Mistry, B.V. Subcellular localization of the mouse PRAMEL1 and PRAMEX1 reveals multifaceted roles in the nucleus and cytoplasm of germ cells during spermatogenesis. Cell Biosci. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Kern, C.H.; Yang, M.; Liu, W.S. The PRAME family of cancer testis antigens is essential for germline development and gametogenesisdagger. Biol. Reprod. 2021, 105, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Bosurgi, L.; Bernink, J.H.; Cuevas, V.D.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Mari, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef]
- Wang, Z.B.; Liu, D.B.; Yan, Q.M.; Liu, F.B.; Zhan, M.B.; Qi, S.M.; Fang, Q.M.; Yao, L.B.; Wang, W.B.; Zhang, R.B.; et al. Activated AXL Protects Against Hepatic Ischemia-reperfusion Injury by Upregulating SOCS-1 Expression. Transplantation 2022. Publish Ah. [Google Scholar] [CrossRef]
- Sang, Y.B.; Kim, J.-H.; Kim, C.-G.; Hong, M.H.; Kim, H.R.; Cho, B.C.; Lim, S.M. The Development of AXL Inhibitors in Lung Cancer: Recent Progress and Challenges. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Duan, Z.; Ho, M. T-Cell Receptor Mimic Antibodies for Cancer Immunotherapy. Mol. Cancer Ther. 2021, 20, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Liu, C.; Wu, Q.; Su, P.; Wu, D.; Guo, J.; Zhou, W.; Xu, Y.; Shi, L.; et al. MSX2 Initiates and Accelerates Mesenchymal Stem/Stromal Cell Specification of hPSCs by Regulating TWIST1 and PRAME. Stem Cell Rep. 2018, 11, 497–513. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedrich, V.; Breitenecker, K.; Ortmayr, G.; Pupp, F.; Huber, H.; Chen, D.; Sahoo, S.; Jolly, M.K.; Mikulits, W. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers 2023, 15, 2415. https://doi.org/10.3390/cancers15092415
Hedrich V, Breitenecker K, Ortmayr G, Pupp F, Huber H, Chen D, Sahoo S, Jolly MK, Mikulits W. PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers. 2023; 15(9):2415. https://doi.org/10.3390/cancers15092415
Chicago/Turabian StyleHedrich, Viola, Kristina Breitenecker, Gregor Ortmayr, Franziska Pupp, Heidemarie Huber, Doris Chen, Sarthak Sahoo, Mohit Kumar Jolly, and Wolfgang Mikulits. 2023. "PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma" Cancers 15, no. 9: 2415. https://doi.org/10.3390/cancers15092415
APA StyleHedrich, V., Breitenecker, K., Ortmayr, G., Pupp, F., Huber, H., Chen, D., Sahoo, S., Jolly, M. K., & Mikulits, W. (2023). PRAME Is a Novel Target of Tumor-Intrinsic Gas6/Axl Activation and Promotes Cancer Cell Invasion in Hepatocellular Carcinoma. Cancers, 15(9), 2415. https://doi.org/10.3390/cancers15092415