

Infrequent Presentations of Chronic NPM1-Mutated Myeloid Neoplasms: Clinicopathological Features of Eight Cases from a Single Institution and Review of the Literature

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
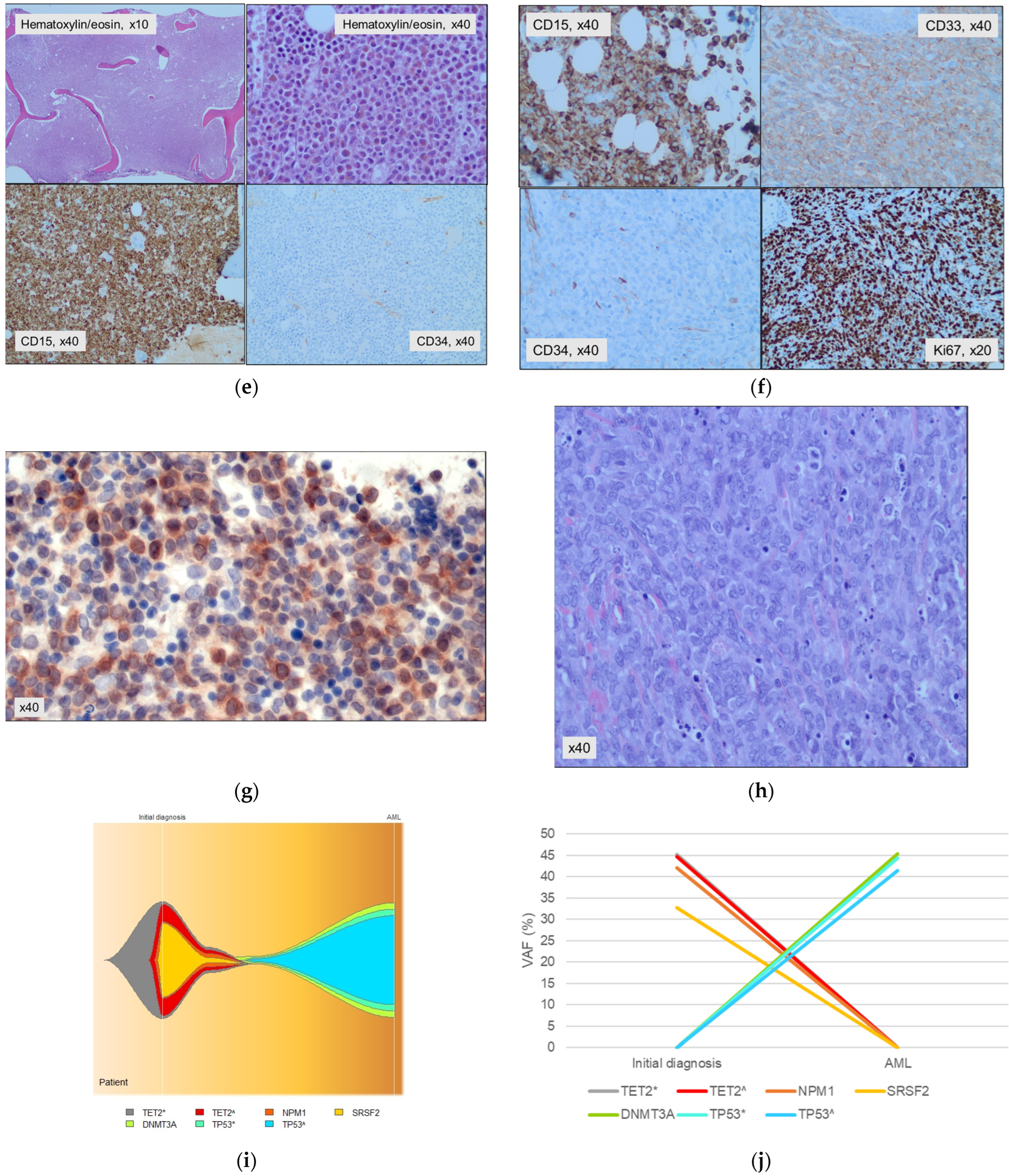
2. An Atypical Presentation of NPM1mut MN
3. Seven Additional Cases of Unusual Presentation of NPM1mut MN
4. Review
4.1. NPM1 in Leukemogenesis and Its Clinical Impact
4.2. NPM1 Mutation in CMML
4.3. NPM1 Mutation in MDS
4.4. NPM1 Mutation in Other Non-Acute Myeloid Neoplasms
5. Conclusions and Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, P.P.; Aarabi, M.; Aggarwal, N. Genomics of myelodysplastic/myeloproliferative neoplasm. Semin. Diagn. Pathol. 2023, 40, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hasserjian, R.P.; Orazi, A.; Orfao, A.; Rozman, M.; Wang, S.A. The International Consensus Classification of myelodysplastic syndromes and related entities. Virchows Arch. 2023, 482, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Ganan-Gomez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; DiNardo, C.; Borthakur, G.; et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019, 3, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Ho, C.; Ptashkin, R.N.; Sadigh, S.; Bagg, A.; Geyer, J.T.; Xu, M.L.; Prebet, T.; Mason, E.F.; Seegmiller, A.C.; et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv. 2019, 3, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Nasillo, V.; Paolini, A.; Bettelli, F.; Pioli, V.; Giusti, D.; Gilioli, A.; Colasante, C.; Acquaviva, G.; Riva, G.; et al. NPM1-Mutated Myeloid Neoplasms with <20% Blasts: A Really Distinct Clinico-Pathologic Entity? Int. J. Mol. Sci. 2020, 21, 8975. [Google Scholar] [PubMed]
- Prakash, S.; Arber, D.A.; Bueso-Ramos, C.; Hasserjian, R.P.; Orazi, A. Advances in myelodysplastic/myeloproliferative neoplasms. Virchows Arch. 2023, 482, 69–83. [Google Scholar] [CrossRef]
- Matanes, F.; AbdelAzeem, B.M.A.; Shah, G.; Reddy, V.; Saad, A.; Papadantonakis, N. Chronic myelomonocytic leukemia associated with myeloid sarcomas and NPM1 mutation: A case report and literature review. Ther. Adv. Hematol. 2019, 10, 2040620719854596. [Google Scholar] [CrossRef]
- Falini, B. NPM1-mutated acute myeloid leukemia: New pathogenetic and therapeutic insights and open questions. Am. J. Hematol. 2023, 98, 1452–1464. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P.; Brunetti, L.; Gjertsen, B.T.; Andresen, V. The NPM1 mutant defines AML irrespective of blast count. Am. J. Hematol. 2023, 98, E187–E189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Campion, V.; Dickson, M.; Tang, C. Acute myeloid leukaemia with NPM1 mutation: No longer having an absolute blast (count). Pathology 2023, 55, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Hasserjian, R.P.; Döhner, H. Distinguishing AML from MDS: A fixed blast percentage may no longer be optimal. Blood 2022, 139, 323–332. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Garcia-Manero, G.; Kantarjian, H.M. Time to blur the blast boundaries. Cancer 2022, 128, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, X.; Xu, F.; Wu, D.; He, Q.; Song, L.; Xiao, C.; Zhao, Y.; Zhang, Z.; Guo, J.; et al. NPM1 mutation with DNMT3A wild type defines a subgroup of MDS with particularly favourable outcomes after decitabine therapy. Br. J. Haematol. 2020, 189, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Wong, C.Y.G.; Gill, H. Targeting and Monitoring Acute Myeloid Leukaemia with Nucleophosmin-1 (NPM1) Mutation. Int. J. Mol. Sci. 2023, 24, 3161. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Díez, S.; Zugasti, I.; Calvo, X.; Schulz, F.; Pita, A.A.; Mora, E.; Falantes, J.F.; Azaceta, G.; Ibáñez, M.; Chen, T.; et al. Chronic Myelomonocytic Leukemia (CMML) with AML Typical Mutations (NPM1, FLT3 or CEBPA) Identify a High-Risk CMML Group Independent of Molecular-Cpss. Blood 2023, 142, 3231. [Google Scholar] [CrossRef]
- Castaño-Díez, S.; López-Guerra, M.; Bosch-Castañeda, C.; Bataller, A.; Charry, P.; Esteban, D.; Guijarro, F.; Jiménez-Vicente, C.; Castillo-Girón, C.; Cortes, A.; et al. Real-World Data on Chronic Myelomonocytic Leukemia: Clinical and Molecular Characteristics, Treatment, Emerging Drugs, and Patient Outcomes. Cancers 2022, 14, 4107. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Martinelli, P.; Zamponi, R.; Shing, D.C.; Bonetti, P.; Luzi, L.; Volorio, S.; Bernard, L.; Pruneri, G.; Alcalay, M.; et al. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006, 66, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.; Davoli, T.; Sironi, C.; Amati, B.; Pelicci, P.G.; Colombo, E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J. Cell Biol. 2008, 182, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wanzel, M.; Russ, A.C.; Kleine-Kohlbrecher, D.; Colombo, E.; Pelicci, P.G.; Eilers, M. A ribosomal protein L23-nucleophosmin circuit coordinates Mizl function with cell growth. Nat. Cell Biol. 2008, 10, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ebrahem, Q.; Mahfouz, R.Z.; Hasipek, M.; Enane, F.; Radivoyevitch, T.; Rapin, N.; Przychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Investig. 2018, 128, 4260–4279. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, M.; Tan, C.H.; Ng, R.; Zeiger, L.; Lau, J.; Lee, J.; Dey, A.; Philp, R.; Li, Q.; Lim, T.M.; et al. Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J. Mol. Biol. 2008, 378, 302–317. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell. 2018, 34, 499–512.e9. [Google Scholar] [CrossRef]
- Uckelmann, H.J.; Haarer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; Perner, F.; Rajput, M.; Antonissen, N.J.C.; Wen, Y.; et al. Mutant NPM1 Directly Regulates Oncogenic Transcription in Acute Myeloid Leukemia. Cancer Discov. 2023, 13, 746–765. [Google Scholar] [CrossRef]
- Kaseb, H.; Visconte, V.; Socha, D.S.; Crane, G.M.; Durkin, L.; Cook, J.R.; Maciejewski, J.P.; Hsi, E.D.; Rogers, H.J. The clinicopathologic significance of NPM1 mutation and ability to detect mutated NPM1 by immunohistochemistry in non-AML myeloid neoplasms. Genes Chromosomes Cancer 2023, 62, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Paolini, A.; Morselli, M.; Bigliardi, S.; Bonacorsi, G.; Leonardi, G.; Coluccio, V.; Maccaferri, M.; Fantuzzi, V.; Faglioni, L.; et al. NPM1 mutations may reveal acute myeloid leukemia in cases otherwise morphologically diagnosed as myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Leuk Lymphoma 2015, 56, 3222–3226. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016, 6, e393. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Duchmann, M.; Lucas, N.; Solary, E. CMML: Clinical and molecular aspects. Int. J. Hematol. 2017, 105, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P.; Bowen, D.; Cross, N.C.P.; Cortes, J.; De Witte, T.; Germing, U.; Onida, F.; Padron, E.; Platzbecker, U.; et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations From the European Hematology Association and the European LeukemiaNet. Hemasphere 2018, 2, e150. [Google Scholar] [CrossRef]
- Elena, C.; Galli, A.; Such, E.; Meggendorfer, M.; Germing, U.; Rizzo, E.; Cervera, J.; Molteni, E.; Fasan, A.; Schuler, E.; et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood 2016, 128, 1408–1417. [Google Scholar] [CrossRef]
- Duchmann, M.; Yalniz, F.F.; Sanna, A.; Sallman, D.; Coombs, C.C.; Renneville, A.; Kosmider, O.; Braun, T.; Platzbecker, U.; Willems, L.; et al. Prognostic Role of Gene Mutations in Chronic Myelomonocytic Leukemia Patients Treated with Hypomethylating Agents. EBioMedicine 2018, 31, 174–181. [Google Scholar] [CrossRef]
- Kuo, M.C.; Liang, D.C.; Huang, C.F.; Shih, Y.S.; Wu, J.H.; Lin, T.L.; Shih, L.Y. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009, 23, 1426–1431. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Knudson, R.A.; Ketterling, R.P.; Tefferi, A.; Solary, E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: A two-center study of 466 patients. Leukemia 2014, 28, 2206–2212. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Wassie, E.A.; Padron, E.; Onida, F.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; et al. Chronic myelomonocytic leukemia in younger patients: Molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J. 2015, 5, e270. [Google Scholar] [CrossRef] [PubMed]
- Shou, L.H.; Cao, D.; Dong, X.H.; Fang, Q.; Wu, Y.; Zhang, Y.; Fei, J.P.; Xu, B.L. Prognostic significance of SETBP1 mutations in myelodysplastic syndromes, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia: A meta-analysis. PLoS ONE 2017, 12, e0171608. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.M.; Kim, S.M.; Nam, Y.; Kim, J.; Kim, S.; Ahn, Y.O.; Park, Y.; Yoon, S.S.; Shin, S.; Kwon, S.; et al. Targeted sequencing aids in identifying clonality in chronic myelomonocytic leukemia. Leuk Res. 2019, 84, 106190. [Google Scholar] [CrossRef] [PubMed]
- Caudill, J.S.; Sternberg, A.J.; Li, C.Y.; Tefferi, A.; Lasho, T.L.; Steensma, D.P. C-terminal nucleophosmin mutations are uncommon in chronic myeloid disorders. Br. J. Haematol. 2006, 133, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Jelinek, J.; Beran, M.; Verstovsek, S.; Kantarjian, H.M.; Issa, J.P. Mutations and promoter methylation status of NPM1 in myeloproliferative disorders. Haematologica 2006, 91, 1147–1148. [Google Scholar] [PubMed]
- Courville, E.L.; Wu, Y.; Kourda, J.; Roth, C.G.; Brockmann, J.; Muzikansky, A.; Fathi, A.T.; de Leval, L.; Orazi, A.; Hasserjian, R.P. Clinicopathologic analysis of acute myeloid leukemia arising from chronic myelomonocytic leukemia. Mod. Pathol. 2013, 26, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zuo, Z.; Fu, B.; Oki, Y.; Tang, G.; Goswami, M.; Priyanka, P.; Muzzafar, T.; Medeiros, L.J.; Luthra, R.; et al. Chronic myelomonocytic leukemia with nucleophosmin (NPM1) mutation. Eur. J. Haematol. 2016, 96, 65–71. [Google Scholar] [CrossRef]
- Vallapureddy, R.; Lasho, T.L.; Hoversten, K.; Finke, C.M.; Ketterling, R.; Hanson, C.; Gangat, N.; Tefferi, A.; Patnaik, M.M. Nucleophosmin 1 (NPM1) mutations in chronic myelomonocytic leukemia and their prognostic relevance. Am. J. Hematol. 2017, 92, E614–E618. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.; Zoi, K.; Waghorn, K.; Hidalgo-Curtis, C.; Score, J.; Jones, A.; Grand, F.; Reiter, A.; Hochhaus, A.; et al. Transcription factor mutations in myelodysplastic/myeloproliferative neoplasms. Haematologica 2010, 95, 1473–1480. [Google Scholar] [CrossRef]
- Nie, Y.; Shao, L.; Zhang, H.; He, C.K.; Li, H.; Zou, J.; Chen, L.; Ji, H.; Tan, H.; Lin, Y.; et al. Mutational landscape of chronic myelomonocytic leukemia in Chinese patients. Exp. Hematol. Oncol. 2022, 11, 32. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Yang, L.; Xiao, Z. NPM1 mutations in myelodysplastic syndromes and acute myeloid leukemia with normal karyotype. Leuk Res. 2007, 31, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Bains, A.; Luthra, R.; Medeiros, L.J.; Zuo, Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: Frequency and potential value for predicting progression to acute myeloid leukemia. Am. J. Clin. Pathol. 2011, 135, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Grisendi, S.; Bernardi, R.; Rossi, M.; Cheng, K.; Khandker, L.; Manova, K.; Pandolfi, P.P. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 2005, 437, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Grisendi, S.; Majid, S.M.; Cheng, K.; Clohessy, J.G.; Viale, A.; Teruya-Feldstein, J.; Pandolfi, P.P. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood 2008, 111, 3859–3862. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Ko, B.S.; Chiou, J.S.; Hsu, Y.C.; Tsai, M.H.; Chiu, Y.C.; Yu, I.S.; Lin, S.W.; Hou, H.A.; Kuo, Y.Y.; et al. A knock-in Npm1 mutation in mice results in myeloproliferation and implies a perturbation in hematopoietic microenvironment. PLoS ONE 2012, 7, e49769. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Sportoletti, P.; Ito, K.; Clohessy, J.G.; Teruya-Feldstein, J.; Kutok, J.L.; Pandolfi, P.P. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 2010, 115, 3341–3345. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Payne, E.M.; Grabher, C.; Lee, J.-S.; Johnston, A.B.; Falini, B.; Kanki, J.P.; Look, A.T. Expression of the cytoplasmic NPM1 mutant (NPMc+) causes the expansion of hematopoietic cells in zebrafish. Blood 2010, 115, 3329–3340. [Google Scholar] [CrossRef]
- Dunbar, A.J.; Rampal, R.K.; Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Alpermann, T.; Dicker, F.; Sundermann, J.; Kern, W.; Haferlach, T. Characterization of NPM1-mutated AML with a history of myelodysplastic syndromes or myeloproliferative neoplasms. Leukemia 2011, 25, 615–621. [Google Scholar] [CrossRef]
- Wang, M.; He, N.; Tian, T.; Liu, L.; Yu, S.; Ma, D. Mutation analysis of JAK2V617F, FLT3-ITD, NPM1, and DNMT3A in Chinese patients with myeloproliferative neoplasms. Biomed. Res. Int. 2014, 2014, 485645. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. World Health Organization Classification of Tumours: Tumours of Haematopoietic and Lymphoid Tissue, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2008. [Google Scholar]
- Gotlib, J. Molecular Classification and Pathogenesis of Eosinophilic Disorders: 2005 Update. Acta Haematol. 2005, 114, 7–25. [Google Scholar] [CrossRef]
- Helbig, G.; Soja, A.; Bartkowska-Chrobok, A.; Kyrcz-Krzemień, S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am. J. Hematol. 2012, 87, 643–645. [Google Scholar] [CrossRef]
- Hofmans, M.; Delie, A.; Vandepoele, K.; Van Roy, N.; Van der Meulen, J.; Philippe, J.; Moors, I. A case of chronic eosinophilic leukemia with secondary transformation to acute myeloid leukemia. Leuk Res. Rep. 2018, 9, 45–47. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patient ID (Age, Yrs/Sex) | ICC and WHO 2022 Classification | WBC (×109/L) | PLT (×109/L) | Hb (g/L) | BMB (%) | Cytology * | Karyotype | Mutations Detected | Treatment for mutNPM1-MN and Response | Status at Last Follow-Up (Survival in Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| NPM1mut-CMML | ||||||||||
| 55/F | CMML-2 ^ | 14.95 | 98 | 98 | 10–15 (BM biopsy) | Granulocytic dysplasia | 46, XX [18] | NPM1, CEBPA, DNMT3A, FLT3-TKD, and IDH1 | (1) ESA; (2) Idarubicin + Cytarabine + Midostaurin followed by alloHCT (mCR); (3) Sorafenib (mCR) | Dead in remission (28) |
| 56/M | CMML-2 ^ | 12.24 | 70 | 85 | 19 | Three-lineage dysplasia | 46, XY, t(14;15)(q32;q22) [18] | NPM1, FLT3-TKD, FLT3-ITD, and TET2 | (1) Azacytidine x1 (SD); (2)Idarubicin + Cytarabine + Midostaurin followed by HiDAC x2 (CR); (3)Venetoclax + Azacytidine x1 followed by sequential alloHCT (mCR) | Dead with disease (29) |
| 73/F | CMML-2 ^ | 30.60 | 110 | 49 | 16 | Granulocytic dysplasia | 46, XX [18] | NPM1, DNMT3 (2 variants), and FLT3 | Hydroxyurea (PD) | Death with disease (6) |
| 60/M | CMML-2 ^ | 27.93 | 35 | 58 | 13 | Granulocytic dysplasia | 46, XY [18] | NPM1 and DNMT3A (2 variants) | (1) Idarubicin + Cytarabine (mCR); (2) Venetoclax + Azacytidine x12 (mCR) | Alive without disease (39) |
| 80/M | CMML-1 + BPDCN | 15.71 | 101 | 138 | 11 | Three-lineage dysplasia | 45, X, -Y | CALR, MPL, NRAS, PHF6, and TET2 (2 variants) (bone marrow)/NPM1, CALR, NRAS, and TET2 (2 variants) (skin) | Venetoclax + Azacytidine (SD) | Dead with disease (12) |
| NPM1mut-MDS | ||||||||||
| 71/F | MDS/AML, MDS-IB | 2.4 | 146 | 117 | 15 | Granulocytic dysplasia | 46, XX | NPM1 and TET2 | (1) Idarubicin + Cytarabine followed by HiDACx1 (CR); (2)Venetoclax + Azacytidine x12 (mCR) | Alive in remission (17) |
| 84/F | MDS NOS, MDS-LB | 8.47 | 81 | 132 | 4 | Megakaryocytic and granulocytic dysplasia | 46, XX | NPM1, EZH2, TET2, and STAG2 | Transfusion support | Dead with disease (16) |
| Others NPM1mut-MN | ||||||||||
| 63/M | Chronic eosinophilic leukemia vs. AML | 3.25 | 70 | 111 | 0 | Granulocytic dysplasia | NA | NPM1, SRSF2, and TET2 | (1) Idarubicin + Cytarabine followed by HiDAC x1 (CR) and consolidated with alloHCT (mCR); (2) Azacytidine + magrolimab (PD) | Dead with disease (24) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaño-Díez, S.; Guijarro, F.; López-Guerra, M.; Pérez-Valencia, A.I.; Gómez-Núñez, M.; Colomer, D.; Díaz-Beyá, M.; Esteve, J.; Rozman, M. Infrequent Presentations of Chronic NPM1-Mutated Myeloid Neoplasms: Clinicopathological Features of Eight Cases from a Single Institution and Review of the Literature. Cancers 2024, 16, 705. https://doi.org/10.3390/cancers16040705
Castaño-Díez S, Guijarro F, López-Guerra M, Pérez-Valencia AI, Gómez-Núñez M, Colomer D, Díaz-Beyá M, Esteve J, Rozman M. Infrequent Presentations of Chronic NPM1-Mutated Myeloid Neoplasms: Clinicopathological Features of Eight Cases from a Single Institution and Review of the Literature. Cancers. 2024; 16(4):705. https://doi.org/10.3390/cancers16040705
Chicago/Turabian StyleCastaño-Díez, Sandra, Francesca Guijarro, Mònica López-Guerra, Amanda Isabel Pérez-Valencia, Marta Gómez-Núñez, Dolors Colomer, Marina Díaz-Beyá, Jordi Esteve, and María Rozman. 2024. "Infrequent Presentations of Chronic NPM1-Mutated Myeloid Neoplasms: Clinicopathological Features of Eight Cases from a Single Institution and Review of the Literature" Cancers 16, no. 4: 705. https://doi.org/10.3390/cancers16040705
APA StyleCastaño-Díez, S., Guijarro, F., López-Guerra, M., Pérez-Valencia, A. I., Gómez-Núñez, M., Colomer, D., Díaz-Beyá, M., Esteve, J., & Rozman, M. (2024). Infrequent Presentations of Chronic NPM1-Mutated Myeloid Neoplasms: Clinicopathological Features of Eight Cases from a Single Institution and Review of the Literature. Cancers, 16(4), 705. https://doi.org/10.3390/cancers16040705