


Hematological Neoplasms with Eosinophilia

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. The Eosinophil
1.2. Eosinophil Biological Activity
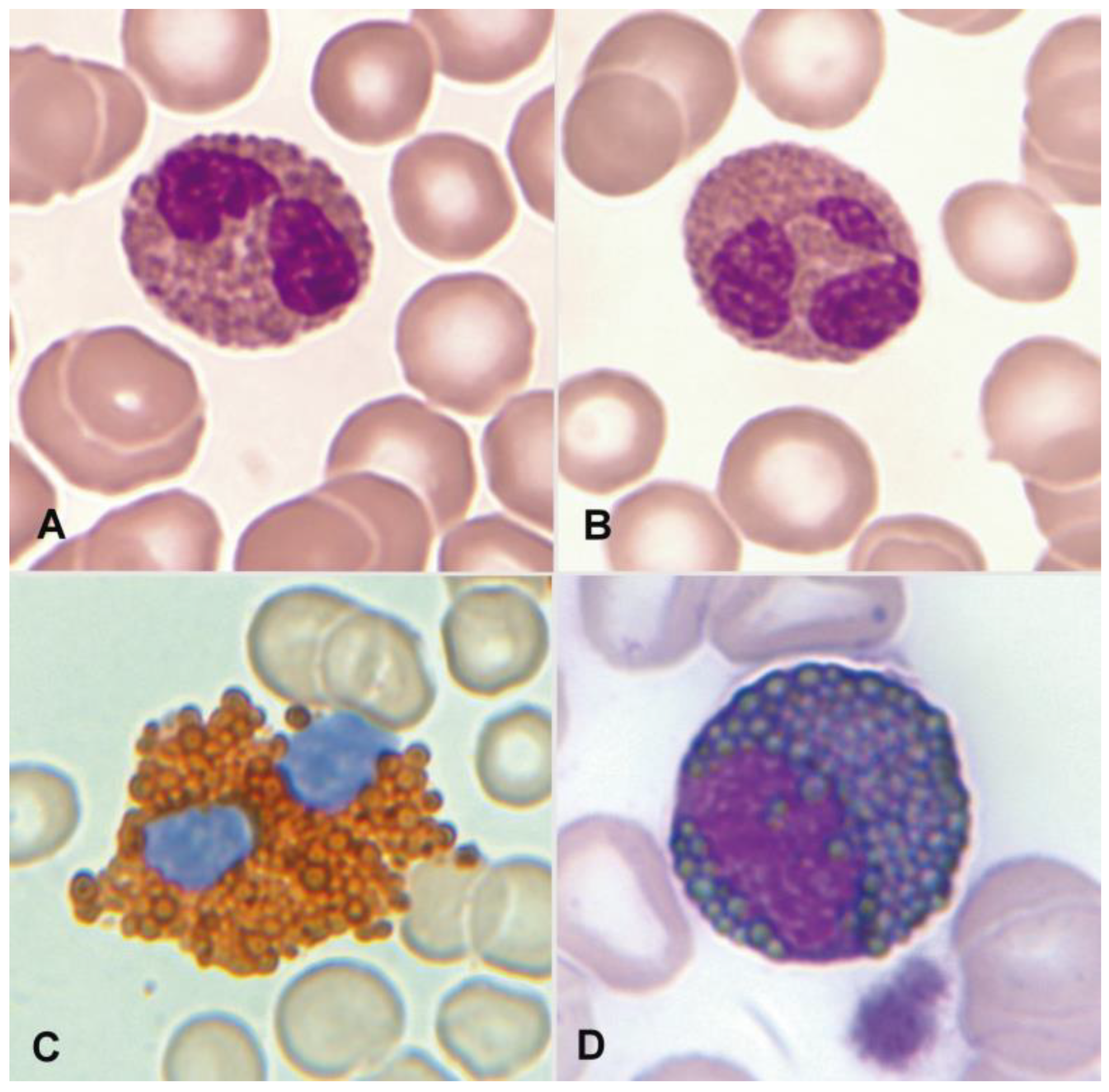
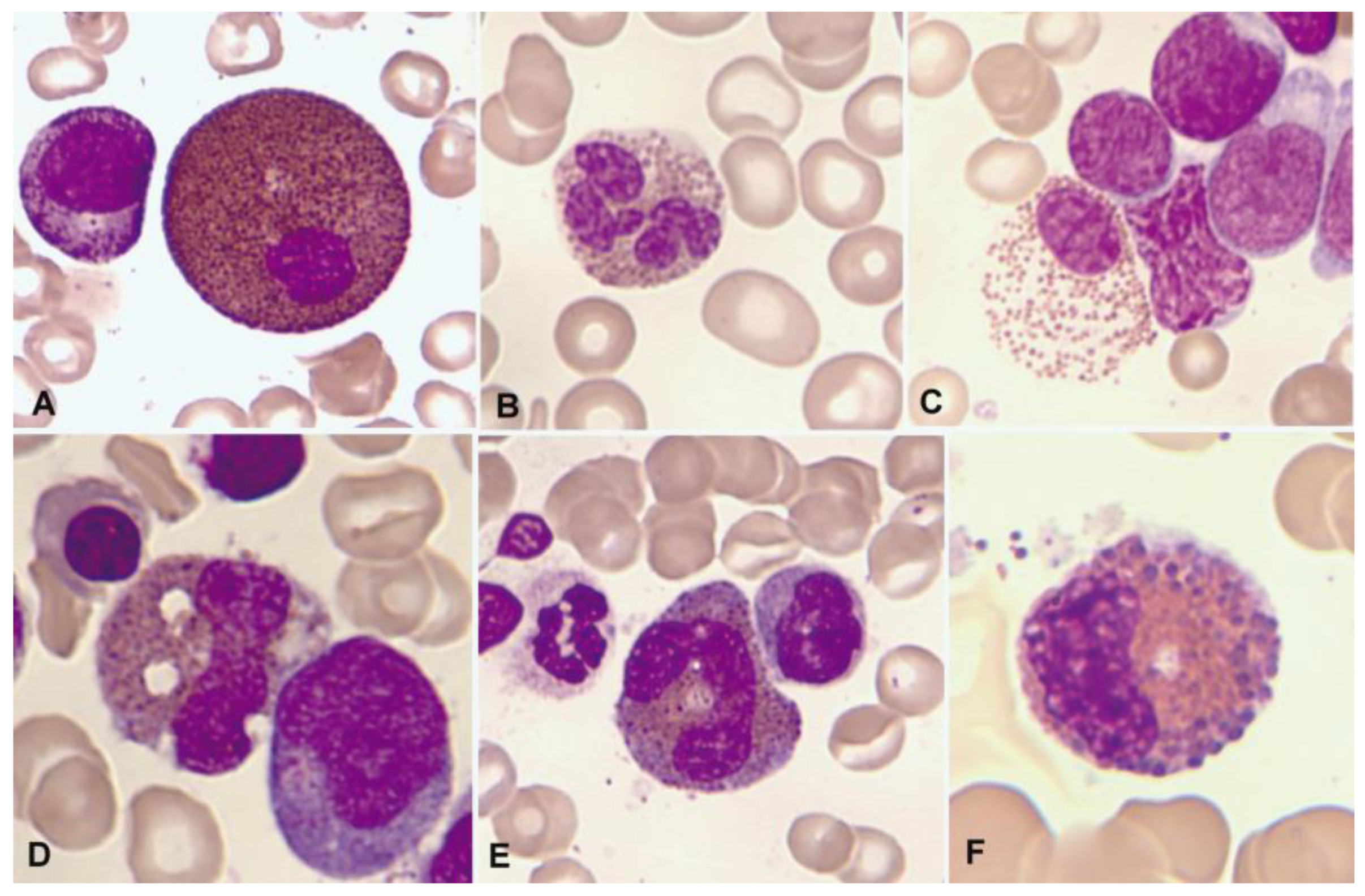
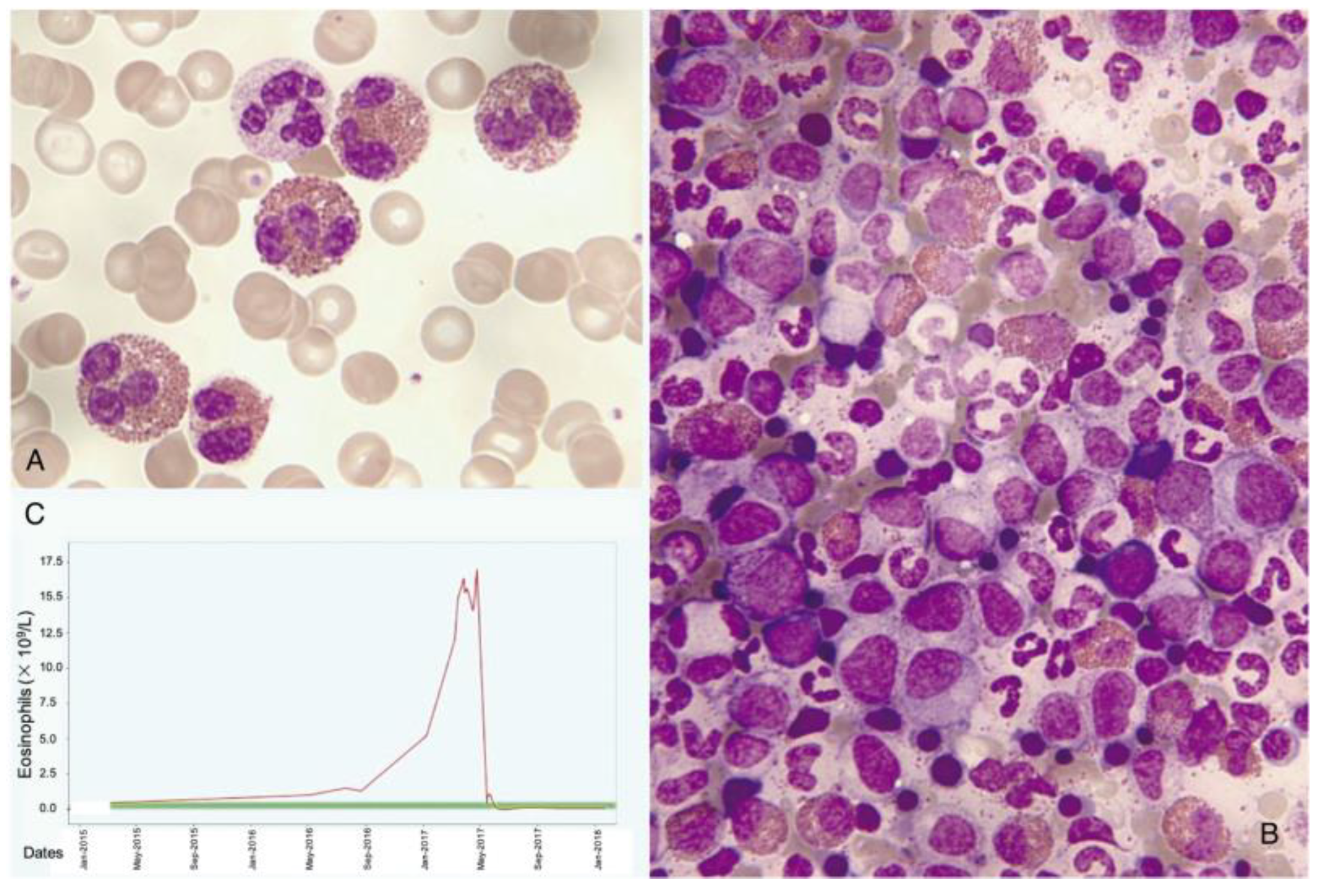
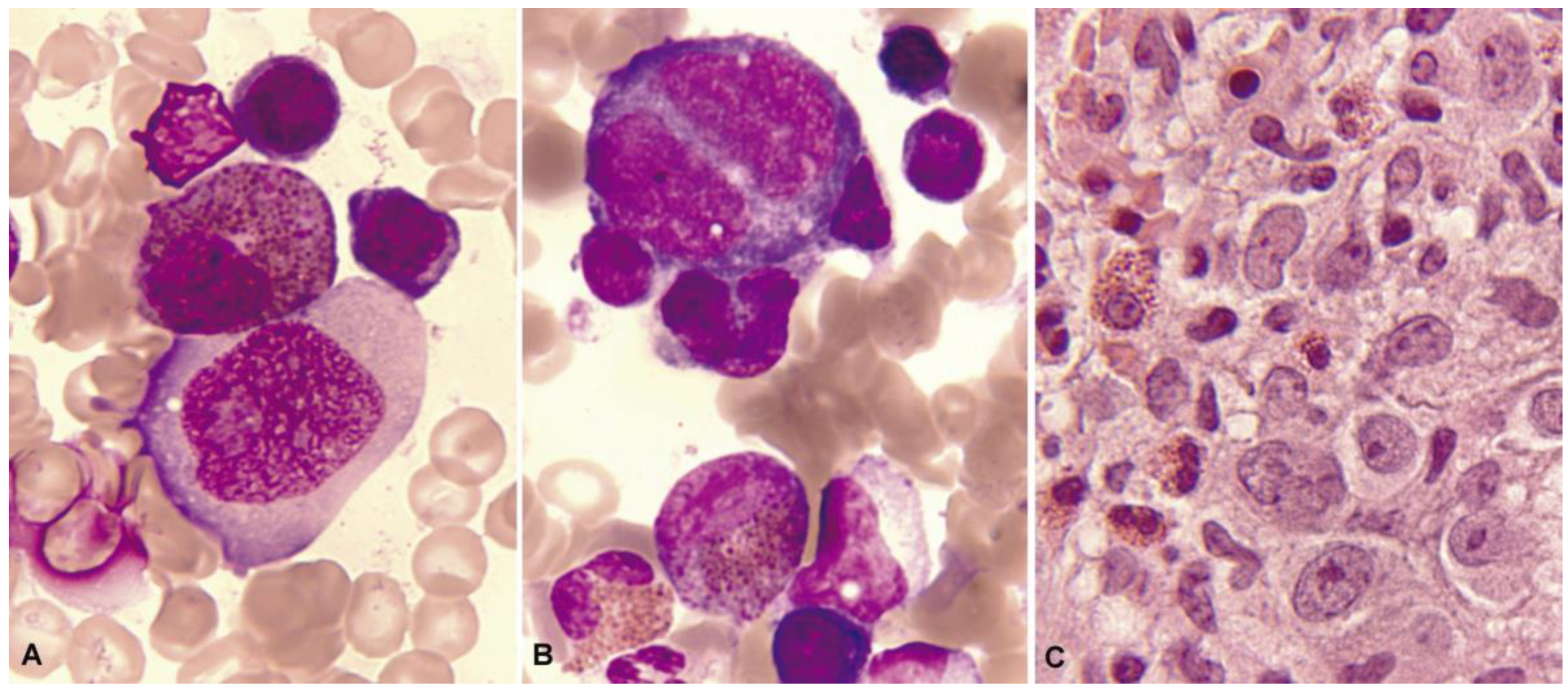
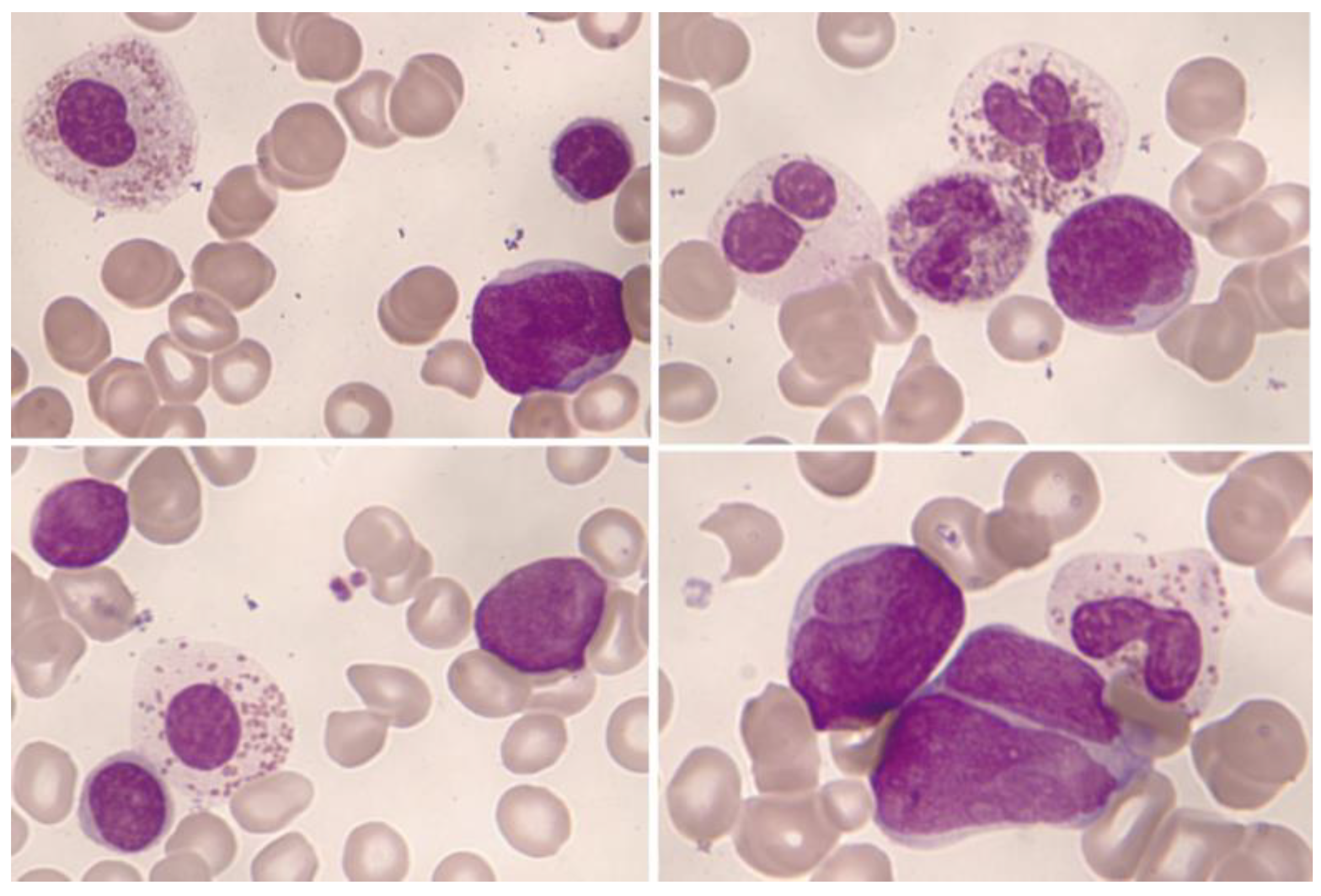
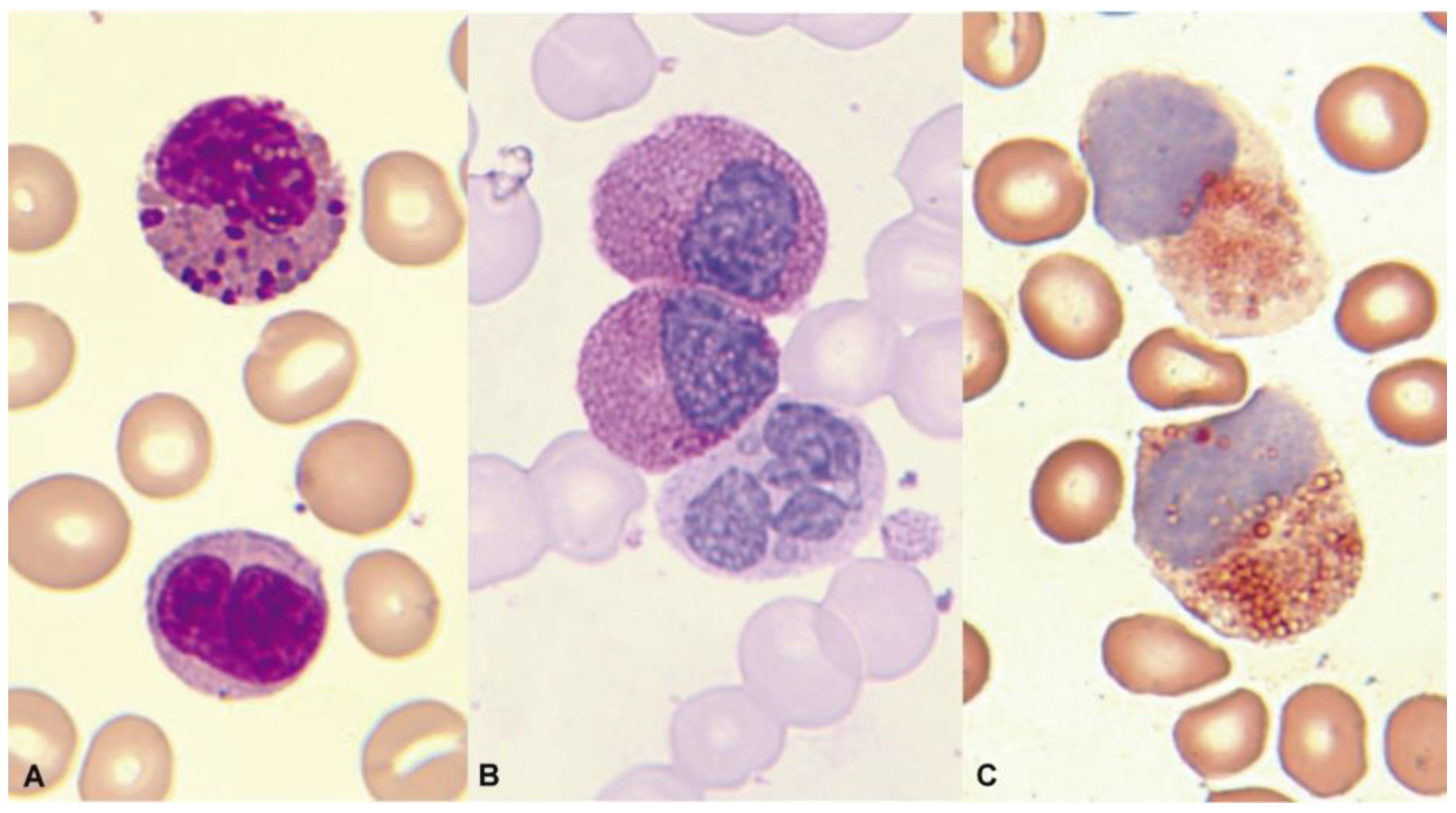
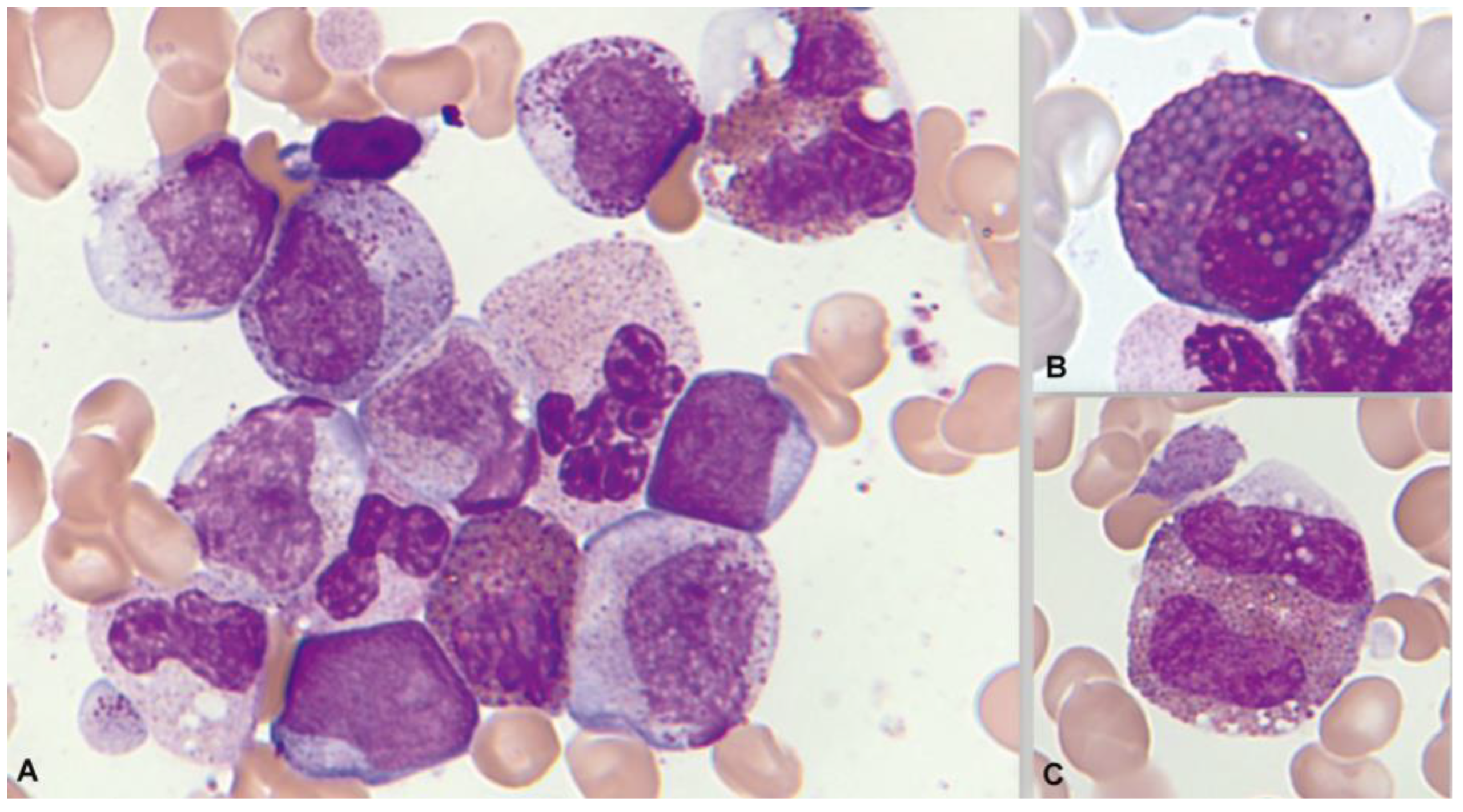
1.3. Normal and Pathological Eosinophil Morphology
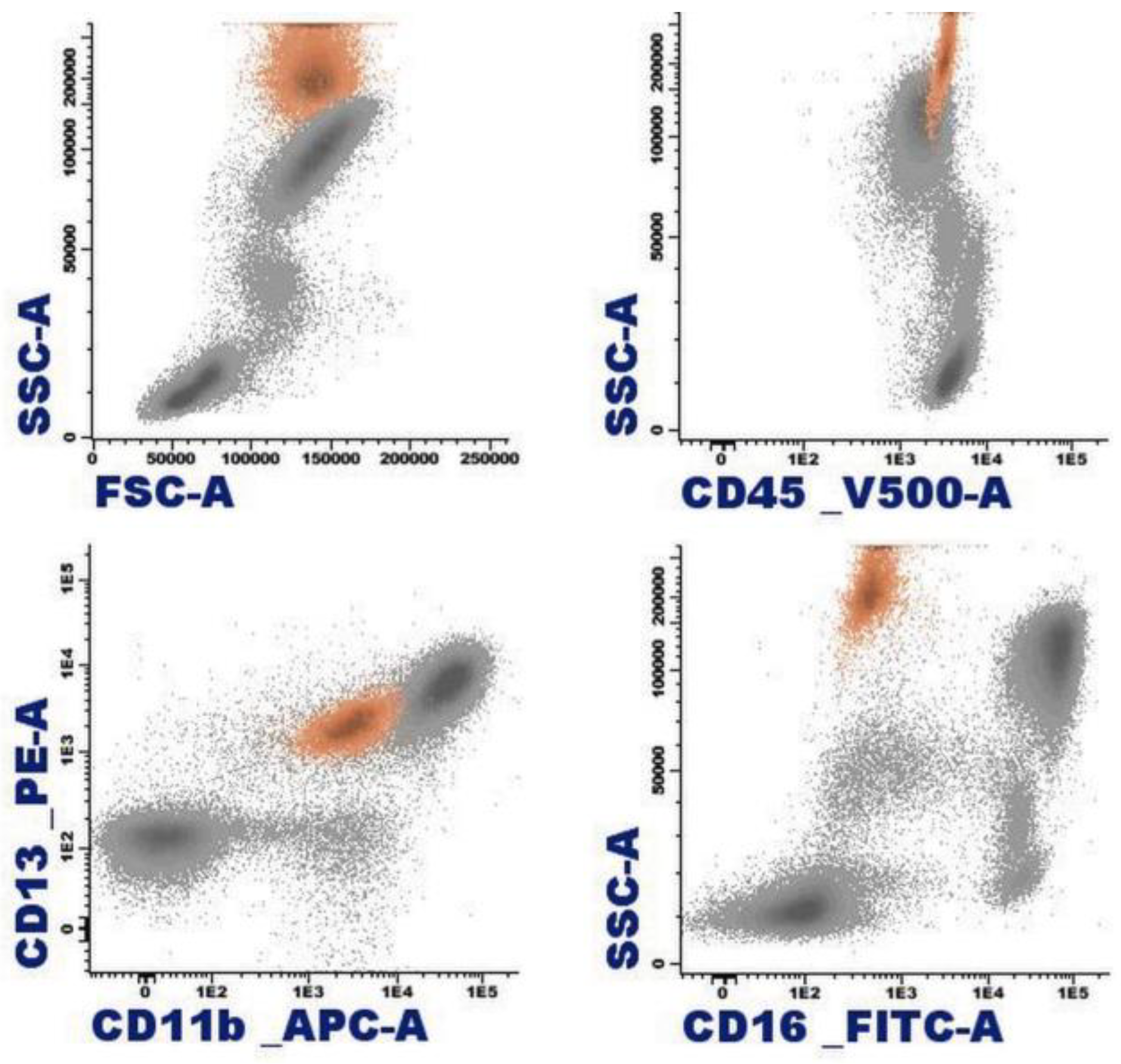
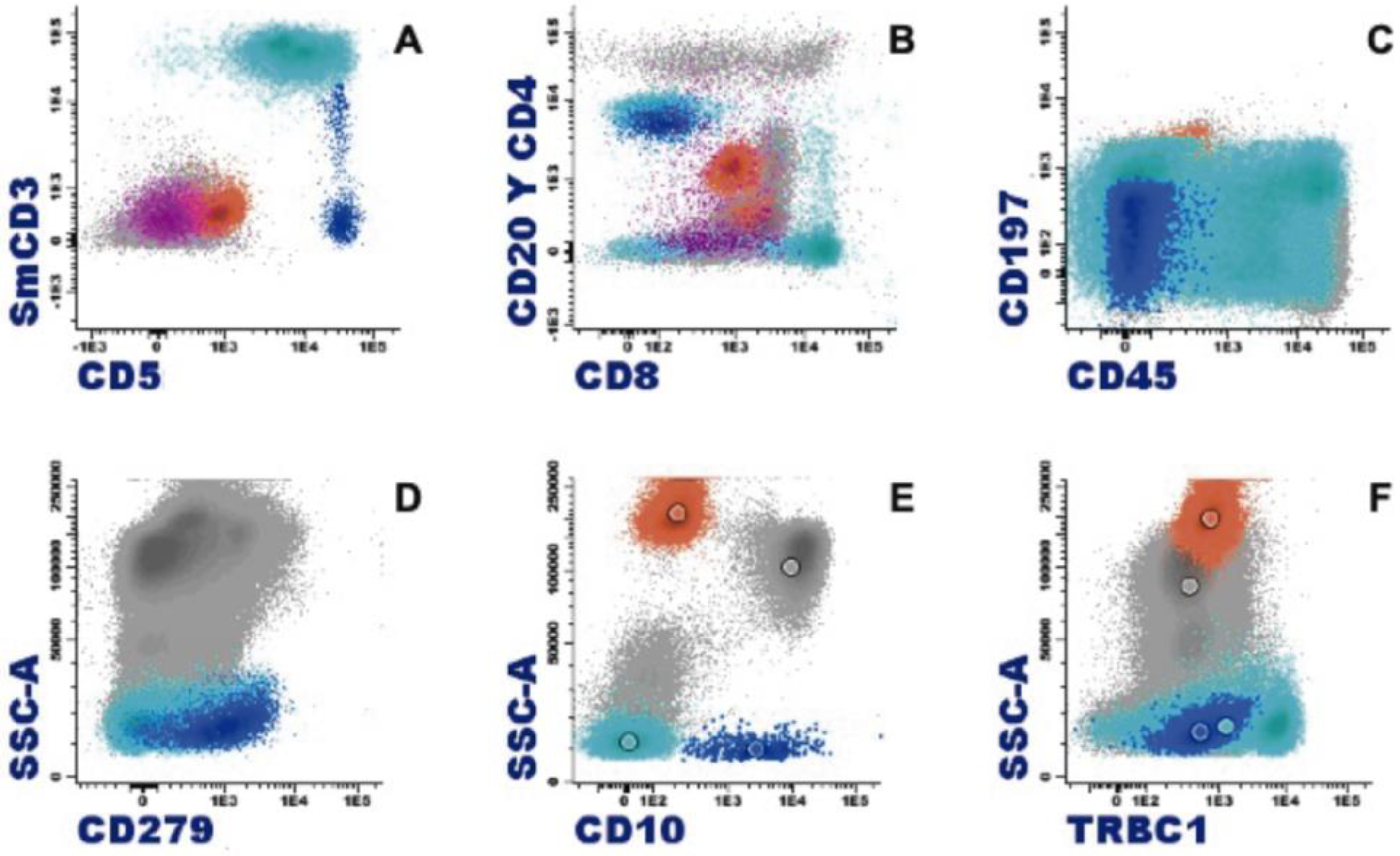
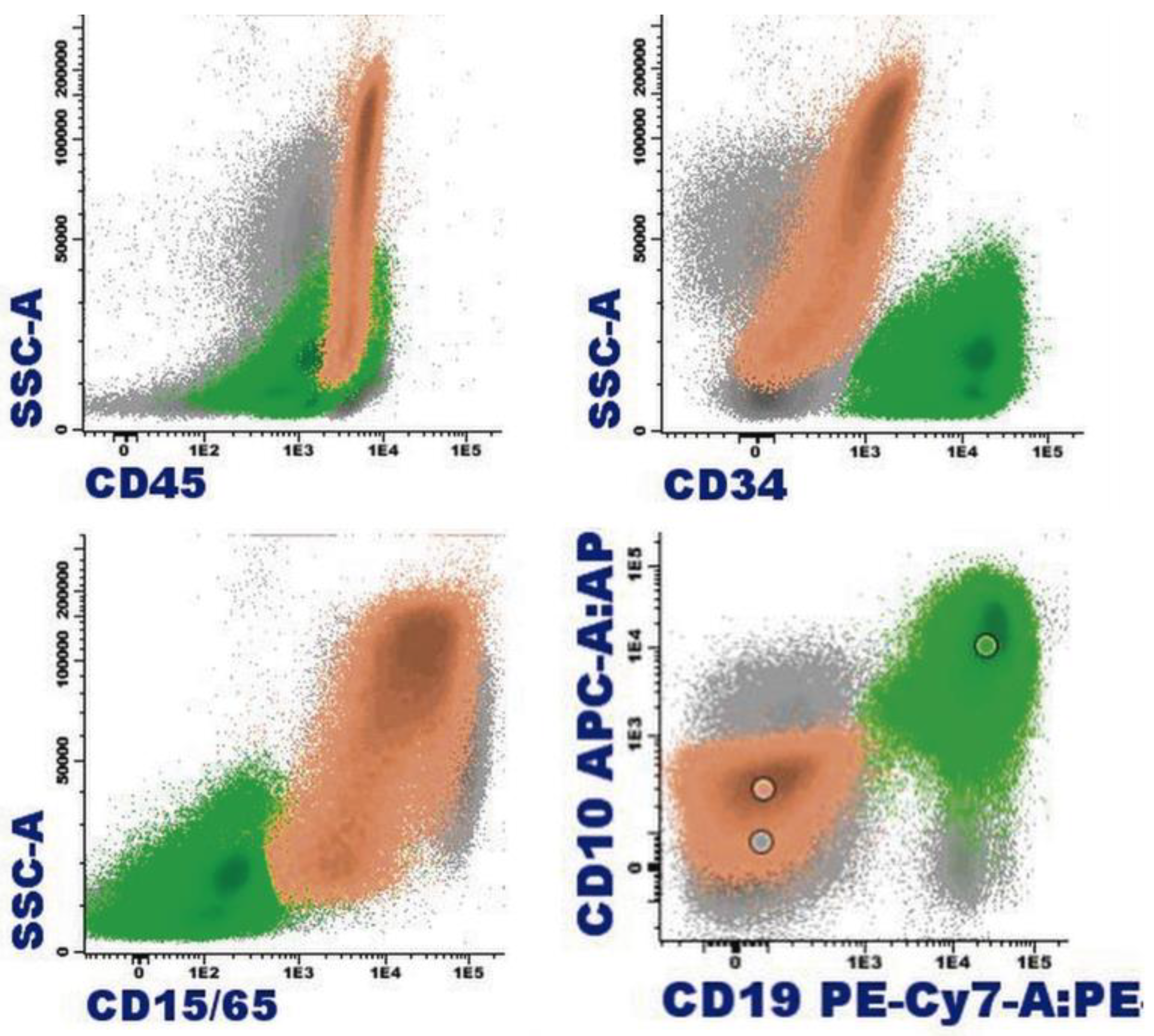
1.4. Eosinophil Flow Cytometry
2. Eosinophilia
3. Hematological Neoplasms Associated with Eosinophilia
3.1. Hematological Neoplasms Associated with Reactive Eosinophilia
3.1.1. Classical Hodgkin Lymphoma
3.1.2. Mature T-Cell Neoplasms
3.1.3. Lymphocytic Variant of Hypereosinophilic Syndrome
3.1.4. B-Lymphoblastic Leukemia/Lymphoma
3.1.5. T-Lymphoblastic Leukemia/Lymphoma
3.2. Hematological Malignancies Associated with Neoplastic or Primary Eosinophilia
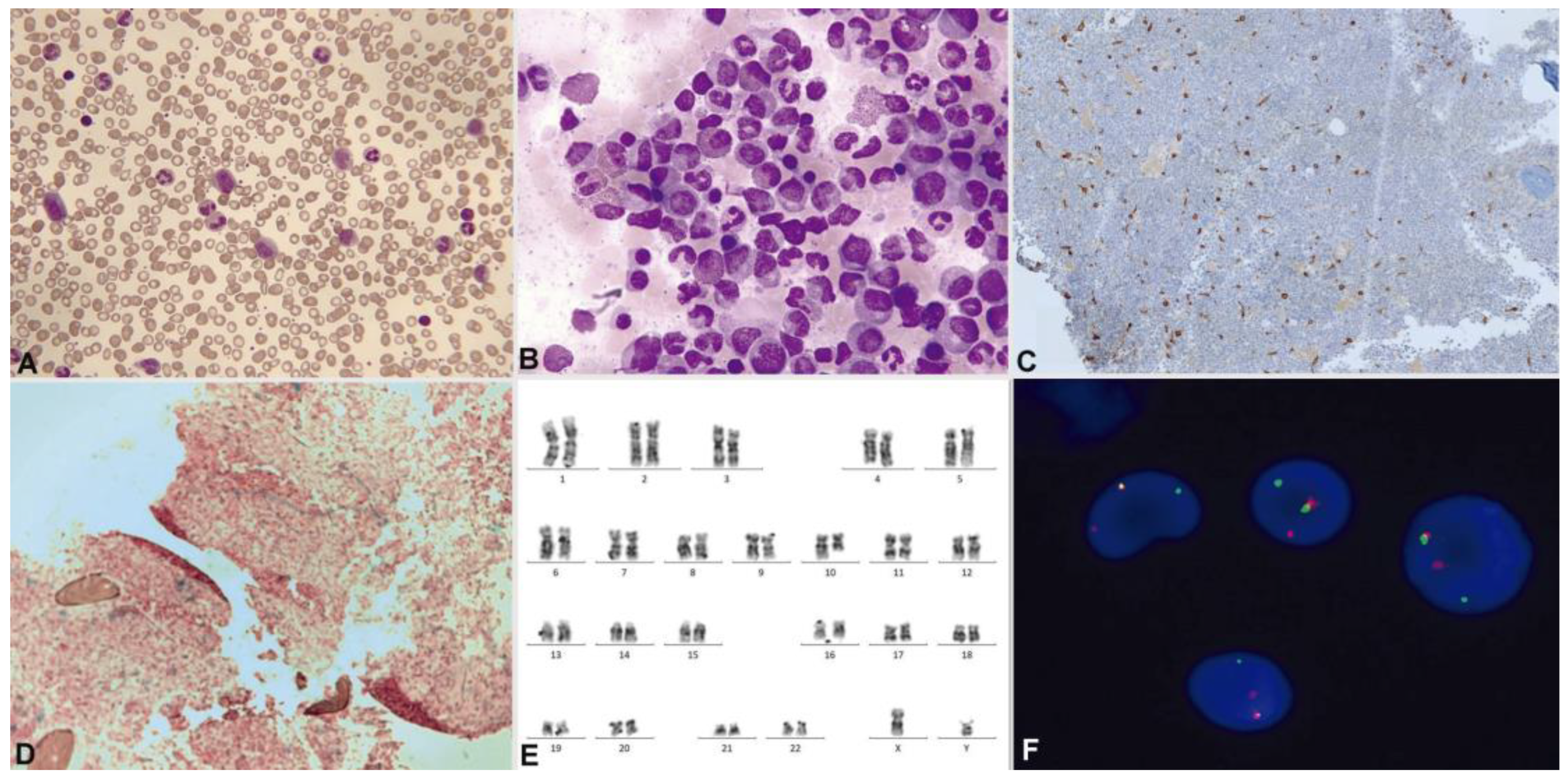
3.2.1. Myeloid/Lymphoid Neoplasms with Eosinophilia and a Defining Gene Rearrangement
Myeloid/Lymphoid Neoplasm with PDGFRA Rearrangement
Myeloid/Lymphoid Neoplasm with PDGFRB Rearrangement
Myeloid/Lymphoid Neoplasm with FGFR1 Rearrangement
Myeloid/Lymphoid Neoplasm with PCM1::JAK2 Rearrangement
Myeloid/Lymphoid Neoplasms with Eosinophilia and Defining Gene Rearrangement: New Entities
- 1.
- Myeloid/lymphoid neoplasms with FLT3/t(v;13q12.2) rearrangements
- 2.
- Myeloid/lymphoid neoplasms with ETV6::ABL1/t(9;12)(q34.1;p13.2) fusion
- 3.
- Myeloid/lymphoid neoplasms with other tyrosine kinase gene fusions
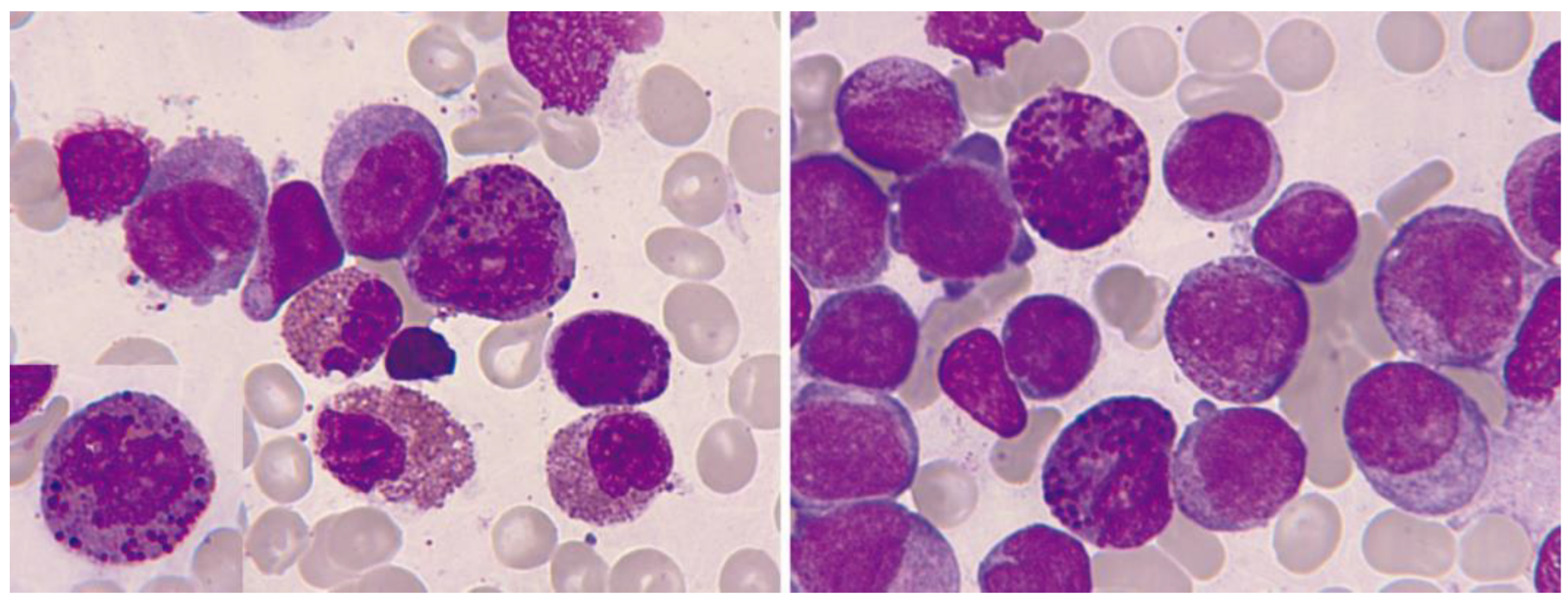
3.2.2. Core Binding Factor Acute Myeloid Leukemias
AML with inv(16)(p13.1q22) o t(16;16)(p13.1;q22); CBFB::MYH11
AML with t(8;21)(q22;q22.1); RUNX1::RUNX1T1
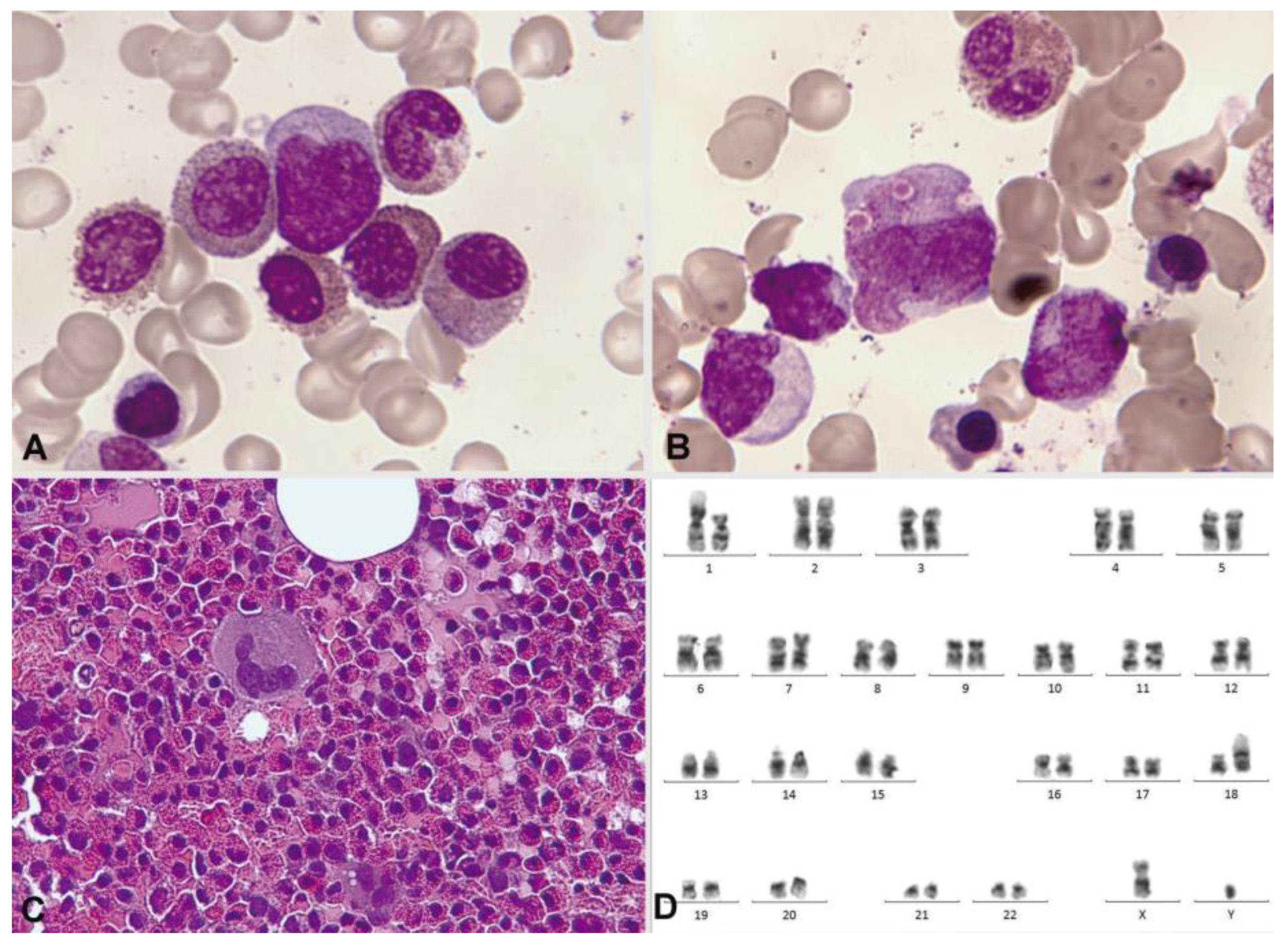
3.2.3. Mastocytosis
3.2.4. Myeloproliferative Neoplasms
Chronic Myeloid Leukemia
Chronic Eosinophilic Leukemia
3.2.5. Myelodysplastic/Myeloproliferative Neoplasms
3.2.6. Myelodysplastic Neoplasms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M/LN-eo with | Frequency * n/Total Cases (%) | Age Median or Range Median (Age Range) | Male/Female Ratio | Most Common Presentation | Treatments | Patient Series |
|---|---|---|---|---|---|---|
| PDGFRA rearrangement | 78/135 (57.8%) | Range median 45.5–51 years (2–82) | 17.8:1 to 43:1 | CEL | Imatinib mesylate | Pozdnyakova et al., 2021 [48] Akiely et al., 2022 [63] Metzgeroth et al., 2023 [49] |
| PDGFRB rearrangement | 26/135 (19.3%) | Range median 38–53 years (0.25–86) | 2.5:1 to 10:1 | MDS/MPN or MPN with eosinophilia | Imatinib mesylate | Metzgeroth et al., 2023 [49] Pozdnyakova et al., 2021 [48] Di Giacomo et al., 2021 [71] Jawhar et al., 2017 [69] Cheah et al., 2014 [70] |
| FGFR1 rearrangement | 9/135 (6.7%) | Range median 46–62 years (0.41–84) | 1.1:1 to 1.6:1 | MPN with/without lymphoblastic lymphoma and eosinophilia | Allogeneic stem cell transplantation Intensive chemotherapy Ponatinib Midastaurina Futibatinib Pemigatinib (NCT03011372) | Patnaik et al., 2010 [142] Strati et al., 2017 [85] Hernández-Boluda et al., 2022 [86] Parasuraman et al., 2022 [87] McKeague et al., 2023 [84] |
| JAK2 rearrangement | 11/135 (8.1%) | Median 47 (6–86) | 3.4:1 | MPN or MDS/MPN with eosinophilia | Allogeneic stem cell transplantation Inh JAK1/JAK2 ruxolitinib (Phase II clinical trial with Ruxolitinib NCT03801434) | Tang et al., 2019 [143] Pozdnyakova et al., 2021 [48] Kaplan et al., 2022 [103] |
| FLT3 rearrangement | Not reported | Median 46.5 (0.6–80) | 1.8:1 | MPN or MDS/MPN with eosinophilia | Allogeneic stem cell transplantation Sorafenib, sunitinib, or gilteritinib | Tang et al., 2021 [106] |
| ETV6::ABL1 rearrangement | 11/135 (8.1%) | Median 51 (24–72) | 2.4:1 | CML-like with eosinophilia | Dasatinib or nilotinib Imatinib mesylate | Zaliova et al., 2016 [116] |
| Other tyrosine kinase gene fusions: FGFR2 LYN NTRK3 ALK RET | Not reported | Few cases | Few cases, but predominates in males | Variable | ETV6::FGFR2: TKI (ponatinib) or FGFR inhibitors ETV6::LYN: Dasatinib ETV6::NTRK3: TRK inhibitor RANBP2::ALK: Crizotinib BCR::RET and FGFR1OP::RET: Sorafenib | Isolated cases |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ehrlich, P. Über die spezifischen Granulationen des Blutes. Arch. Anat. Physiol. 1879, 1, 571–579. [Google Scholar]
- Kay, A.B. The early history of the eosinophil. Clin. Exp. Allergy 2015, 45, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Woessner Casas, S.; Florensa Brich, L. La Citología Óptica en el Diagnóstico Hematológico, 5th ed.; Acción Médica S. A.: Madrid, Spain, 2006; ISBN 84-88336-59-4. [Google Scholar]
- Johnston, L.K.; Bryce, P.J. Understanding Interleukin 33 and Its Roles in Eosinophil Development. Front. Med. 2017, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Lucarini, V.; Marone, G.; Mattei, F.; Marone, G.; Schiavoni, G. Eosinophils: The unsung heroes in cancer? Oncoimmunology 2017, 7, e1393134. [Google Scholar] [CrossRef] [PubMed]
- DiScipio, R.G.; Schraufstatter, I.U. The role of the complement anaphylatoxins in the recruitment of eosinophils. Int. Immunopharmacol. 2007, 7, 1909–1923. [Google Scholar] [CrossRef]
- Melo, R.C.N.; Weller, P.F. Contemporary understanding of the secretory granules in human eosinophils. J. Leukoc. Biol. 2018, 104, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Ackerman, S.J. Eosinophil granule proteins: Form and function. J. Biol. Chem. 2014, 289, 17406–17415. [Google Scholar] [CrossRef]
- Kanda, A.; Yasutaka, Y.; Van Bui, D.; Suzuki, K.; Sawada, S.; Kobayashi, Y.; Asako, M.; Iwai, H. Multiple Biological Aspects of Eosinophils in Host Defense, Eosinophil-Associated Diseases, Immunoregulation, and Homeostasis: Is Their Role Beneficial, Detrimental, Regulator, or Bystander? Biol. Pharm. Bull. 2020, 43, 20–30. [Google Scholar] [CrossRef]
- Goasguen, J.E.; Bennett, J.M.; Bain, B.J.; Brunning, R.; Zini, G.; Vallespi, M.T.; Tomonaga, M.; Locher, C.; International Working Group on Morphology of MDS. The role of eosinophil morphology in distinguishing between reactive eosinophilia and eosinophilia as a feature of a myeloid neoplasm. Br. J. Haematol. 2020, 191, 497–504, Erratum in Br. J. Haematol. 2021, 192, 407. [Google Scholar] [CrossRef]
- Zederbauer, M.; Furtmüller, P.G.; Brogioni, S.; Jakopitsch, C.; Smulevich, G.; Obinger, C. Heme to protein linkages in mammalian peroxidases: Impact on spectroscopic, redox and catalytic properties. Nat. Prod. Rep. 2007, 24, 571–584. [Google Scholar] [CrossRef]
- Orfao, A.; Matarraz, S.; Pérez-Andrés, M.; Almeida, J.; Teodosio, C.; Berkowska, M.A.; van Dongen, J.J.M.; EuroFlow. Immunophenotypic dissection of normal hematopoiesis. J. Immunol. Methods 2019, 475, 112684. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.J. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br. J. Haematol. 1996, 95, 2–9. [Google Scholar] [PubMed]
- Valent, P.; Klion, A.D.; Horny, H.P.; Roufosse, F.; Gotlib, J.; Weller, P.F.; Hellmann, A.; Metzgeroth, G.; Leiferman, K.M.; Arock, M.; et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J. Allergy Clin. Immunol. 2012, 130, 607–612.e9. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Klion, A.D.; Roufosse, F.; Simon, D.; Metzgeroth, G.; Leiferman, K.M.; Schwaab, J.; Butterfield, J.H.; Sperr, W.R.; Sotlar, K.; et al. Proposed refined diagnostic criteria and classification of eosinophil disorders and related syndromes. Allergy 2023, 78, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Degenfeld-Schonburg, L.; Sadovnik, I.; Horny, H.P.; Arock, M.; Simon, H.U.; Reiter, A.; Bochner, B.S. Eosinophils and eosinophil-associated disorders: Immunological, clinical, and molecular complexity. Semin. Immunopathol. 2021, 43, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Serrano, J.; Morales-Camacho, R.M.; Caballero-Velázquez, T.; García-Canale, S.; Vargas, M.T.; Prats-Martín, C. Eosinophils engulfing platelets and with ring-shaped nuclei in nivolumab-associated eosinophilia. Br. J. Haematol. 2020, 188, 812. [Google Scholar] [CrossRef]
- Morales-Camacho, R.M.; Prats-Martín, C. Eosinophils with ring-shaped nuclei in a patient treated with adalimumab. Blood 2019, 133, 101. [Google Scholar] [CrossRef]
- Klion, A.D. Approach to the patient with suspected hypereosinophilic syndrome. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 47–54. [Google Scholar] [CrossRef]
- Khoury, P.; Akuthota, P.; Kwon, N.; Steinfeld, J.; Roufosse, F. HES and EGPA: Two Sides of the Same Coin. Mayo Clin. Proc. 2023, 98, 1054–1070. [Google Scholar] [CrossRef]
- Guenzel, A.J.; Smadbeck, J.B.; Golden, C.L.; Williamson, C.M.; Benevides Demasi, J.C.; Vasmatzis, G.; Pearce, K.E.; Olteanu, H.; Xu, X.; Hoppman, N.L.; et al. Clinical utility of next generation sequencing to detect IGH/IL3 rearrangements [t(5;14)(q31.1;q32.1)] in B-lymphoblastic leukemia/lymphoma. Ann. Diagn. Pathol. 2021, 53, 151761. [Google Scholar] [CrossRef]
- Romagnoli, S.; Bartalucci, N.; Gesullo, F.; Balliu, M.; Bonifacio, S.; Fernandez, A.G.L.; Mannelli, F.; Bolognini, D.; Pelo, E.; Mecucci, C.; et al. Nanopore sequencing for the screening of myeloid and lymphoid neoplasms with eosinophilia and rearrangement of PDGFRα, PDGFRβ, FGFR1 or PCM1-JAK2. Biomark. Res. 2021, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Podvin, B.; Roynard, P.; Boudry, A.; Guermouche, H.; Daudignon, A.; Terriou, L.; Bouabdelli, W.; Salameh, M.; Grardel, N.; Duployez, N.; et al. Whole-genome optical mapping to elucidate myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions. Leuk. Res. 2022, 123, 106972. [Google Scholar] [CrossRef] [PubMed]
- Shomali, W.; Damnernsawad, A.; Theparee, T.; Sampson, D.; Morrow, Q.; Yang, F.; Fernandez-Pol, S.; Press, R.; Zehnder, J.; Tyner, J.W.; et al. A novel activating JAK1 mutation in chronic eosinophilic leukemia. Blood Adv. 2021, 5, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lübke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Yin, C.C.; Tam, W.; Walker, S.M.; Kaur, A.; Ouseph, M.M.; Xie, W.; Weinberg, O.K.; Li, P.; Zuo, Z.; Routbort, M.J.; et al. STAT5B mutations in myeloid neoplasms differ by disease subtypes but characterize a subset of chronic myeloid neoplasms with eosinophilia and/or basophilia. Haematologica 2023. Epub ahead of print. [Google Scholar] [CrossRef]
- Umrau, K.; Naganuma, K.; Gao, Q.; Dogan, A.; Kizaki, M.; Roshal, M.; Liu, Y.; Yabe, M. Activating STAT5B mutations can cause both primary hypereosinophilia and lymphocyte-variant hypereosinophilia. Leuk. Lymphoma 2023, 64, 238–241. [Google Scholar] [CrossRef]
- Sreedharanunni, S.; Jamwal, M.; Balakrishnan, A.; Aravindan, A.V.; Sharma, R.; Singh, N.; Rajpal, S.; Singla, S.; Khadwal, A.R.; Ahluwalia, J.; et al. Chronic eosinophilic leukemia with recurrent STAT5B N642H mutation-An entity with features of myelodysplastic syndrome/myeloproliferative neoplasm overlap. Leuk. Res. 2022, 112, 106753. [Google Scholar] [CrossRef]
- Ding, F.; Wu, C.; Li, Y.; Mukherjee, S.; Ghosh, S.; Arrossi, A.V.; Krishnan, S. A case of hypereosinophilic syndrome with STAT5b N642H mutation. Oxf. Med. Case Rep. 2021, 2021, ocaa129. [Google Scholar] [CrossRef]
- Ma, C.A.; Xi, L.; Cauff, B.; DeZure, A.; Freeman, A.F.; Hambleton, S.; Kleiner, G.; Leahy, T.R.; O’Sullivan, M.; Makiya, M.; et al. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood 2017, 129, 650–653. [Google Scholar] [CrossRef]
- Roufosse, F.; Garaud, S.; de Leval, L. Lymphoproliferative disorders associated with hypereosinophilia. Semin. Hematol. 2012, 49, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Samoszuk, M.; Nansen, L. Detection of interleukin-5 messenger RNA in Reed-Sternberg cells of Hodgkin’s disease with eosinophilia. Blood 1990, 75, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, N.; Lima, M.; Villamor, N.; Morán-Plata, F.J.; Barrena, S.; Mateos, S.; Caldas, C.; Balanzategui, A.; Alcoceba, M.; Domínguez, A.; et al. Anti-TRBC1 Antibody-Based Flow Cytometric Detection of T-Cell Clonality: Standardization of Sample Preparation and Diagnostic Implementation. Cancers 2021, 13, 4379. [Google Scholar] [CrossRef] [PubMed]
- Tancrède-Bohin, E.; Ionescu, M.A.; de La Salmonière, P.; Dupuy, A.; Rivet, J.; Rybojad, M.; Dubertret, L.; Bachelez, H.; Lebbé, C.; Morel, P. Prognostic value of blood eosinophilia in primary cutaneous T-cell lymphomas. Arch. Dermatol. 2004, 140, 1057–1061. [Google Scholar] [CrossRef]
- Pulitzer, M.P.; Horna, P.; Almeida, J. Sézary syndrome and mycosis fungoides: An overview, including the role of immunophenotyping. Cytometry B Clin. Cytom. 2021, 100, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Qiao, J.; Liu, Y.; Cao, X.; Tan, R.; Yan, D.; Wang, X.; Li, J.; Yue, B. Differential diagnosis and identification of prognostic markers for peripheral T-cell lymphoma subtypes based on flow cytometry immunophenotype profiles. Front. Immunol. 2022, 13, 1008695. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T.; Karube, K.; Sakihama, S.; Tsuruta, Y.; Awazawa, R.; Hayashi, M.; Nakada, N.; Matsumoto, H.; Yagi, N.; Ohshiro, K.; et al. A Comprehensive Study of the Immunophenotype and its Clinicopathologic Significance in Adult T-Cell Leukemia/Lymphoma. Mod. Pathol. 2023, 36, 100169. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, C. What we have learned about lymphocytic variant hypereosinophilic syndrome: A systematic literature review. Clin. Immunol. 2022, 237, 108982. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Fournier, B.; Balducci, E.; Duployez, N.; Clappier, E.; Cuccuini, W.; Arfeuille, C.; Caye-Eude, A.; Delabesse, E.; Bottollier-Lemallaz, C.E.; Nebral, K.; et al. B-ALL with t(5;14)(q31;q32); IGH-IL3 Rearrangement and Eosinophilia: A Comprehensive Analysis of a Peculiar IGH-Rearranged B-ALL. Front. Oncol. 2019, 9, 1374. [Google Scholar] [CrossRef]
- McClure, B.J.; Heatley, S.L.; Rehn, J.; Breen, J.; Sutton, R.; Hughes, T.P.; Suppiah, R.; Revesz, T.; Osborn, M.; White, D.; et al. High-risk B-cell acute lymphoblastic leukaemia presenting with hypereosinophilia and acquiring a novel PAX5 fusion on relapse. Br. J. Haematol. 2020, 191, 301–304. [Google Scholar] [CrossRef]
- Kwon, A.; Fuda, F.; Gagan, J.; John, S.; Aquino, V.; Chen, W. Rare circulating lymphoblasts with striking eosinophilia: A rare case of B-lymphoblastic leukemia with PAX5::ZCCHC7. Am. J. Hematol. 2023, 98, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Ferruzzi, V.; Santi, E.; Gurdo, G.; Arcioni, F.; Caniglia, M.; Esposito, S. Acute Lymphoblastic Leukemia with Hypereosinophilia in a Child: Case Report and Literature Review. Int. J. Environ. Res. Public Health 2018, 15, 1169. [Google Scholar] [CrossRef] [PubMed]
- Pierini, V.; Bardelli, V.; Giglio, F.; Arniani, S.; Matteucci, C.; Pellanera, F.; Quintini, M.; Crescenzi, B.; Bruno, A.; Sabattini, E.; et al. A Novel t(5;7)(q31;q21)/CDK6::IL3 in Immature T-cell Acute Lymphoblastic Leukemia With IL3 Expression and Eosinophilia. Hemasphere 2022, 6, e795. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Revised 4th Edition; IARC: Lyon, France, 2017. [Google Scholar]
- Kim, A.S.; Pozdnyakova, O. SOHO State of the Art Updates and Next Questions|Myeloid/Lymphoid Neoplasms with Eosinophilia and Gene Rearrangements: Diagnostic Pearls and Pitfalls. Clin. Lymphoma Myeloma Leuk. 2022, 22, 643–651. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Orazi, A.; Kelemen, K.; King, R.; Reichard, K.K.; Craig, F.E.; Quintanilla-Martinez, L.; Rimsza, L.; George, T.I.; Horny, H.P.; et al. Myeloid/Lymphoid Neoplasms Associated with Eosinophilia and Rearrangements of PDGFRA, PDGFRB, or FGFR1 or With PCM1-JAK2. Am. J. Clin. Pathol. 2021, 155, 160–178. [Google Scholar] [CrossRef]
- Metzgeroth, G.; Steiner, L.; Naumann, N.; Lübke, J.; Kreil, S.; Fabarius, A.; Haferlach, C.; Haferlach, T.; Hofmann, W.K.; Reiter, A.; et al. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions: Reevaluation of the defining characteristics in a registry-based cohort. Leukemia 2023, 37, 1860–1867. [Google Scholar] [CrossRef]
- Saft, L.; Kvasnicka, H.M.; Boudova, L.; Gianelli, U.; Lazzi, S.; Rozman, M. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase fusion genes: A workshop report with focus on novel entities and a literature review including paediatric cases. Histopathology 2023, 83, 829–849. [Google Scholar] [CrossRef]
- Shomali, W.; Gotlib, J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 129–148. [Google Scholar] [CrossRef]
- Rohmer, J.; Couteau-Chardon, A.; Trichereau, J.; Panel, K.; Gesquiere, C.; Ben Abdelali, R.; Bidet, A.; Bladé, J.S.; Cayuela, J.M.; Cony-Makhoul, P.; et al. Epidemiology, clinical picture and long-term outcomes of FIP1L1-PDGFRA-positive myeloid neoplasm with eosinophilia: Data from 151 patients. Am. J. Hematol. 2020, 95, 1314–1323. [Google Scholar] [CrossRef]
- Score, J.; Curtis, C.; Waghorn, K.; Stalder, M.; Jotterand, M.; Grand, F.H.; Cross, N.C. Identification of a novel imatinib responsive KIF5B-PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia 2006, 20, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Curtis, C.; Schnittger, S.; Schultheis, B.; Metzgeroth, G.; Schoch, C.; Lengfelder, E.; Erben, P.; Müller, M.C.; Haferlach, T.; et al. Transient response to imatinib in a chronic eosinophilic leukemia associated with ins(9;4)(q33;q12q25) and a CDK5RAP2-PDGFRA fusion gene. Genes Chromosomes Cancer 2006, 45, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.E.; Grand, F.H.; Musto, P.; Clark, A.; Murphy, J.; Perla, G.; Minervini, M.M.; Stewart, J.; Reiter, A.; Cross, N.C. Two novel imatinib-responsive PDGFRA fusion genes in chronic eosinophilic leukaemia. Br. J. Haematol. 2007, 138, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Hochhaus, A.; Bolufer, P.; Reiter, A.; Fernandez, J.M.; Senent, L.; Cervera, J.; Moscardo, F.; Sanz, M.A.; Cross, N.C. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses BCR to PDGFRA. Hum. Mol. Genet. 2002, 11, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Ali, S.M.; Ohgami, R.S.; Campregher, P.V.; Frampton, G.M.; Yelensky, R.; Elvin, J.A.; Palma, N.A.; Erlich, R.; Vergilio, J.A.; et al. Comprehensive genomic profiling identifies a novel TNKS2-PDGFRA fusion that defines a myeloid neoplasm with eosinophilia that responded dramatically to imatinib therapy. Blood Cancer J. 2015, 5, e278. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Sada, A.; Shimokariya, Y.; Monma, F.; Ohishi, K.; Masuya, M.; Nobori, T.; Matsui, T.; Katayama, N. A novel FOXP1-PDGFRA fusion gene in myeloproliferative neoplasm with eosinophilia. Cancer Genet. 2015, 208, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Muehlbacher, V.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular genetic characterization of myeloid/lymphoid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. Haematologica 2018, 103, e348–e350. [Google Scholar] [CrossRef]
- Jain, N.; Khoury, J.D.; Pemmaraju, N.; Kollipara, P.; Kantarjian, H.; Verstovsek, S. Imatinib therapy in a patient with suspected chronic neutrophilic leukemia and FIP1L1-PDGFRA rearrangement. Blood 2013, 122, 3387–3388. [Google Scholar] [CrossRef]
- Huang, Q.; Snyder, D.S.; Chu, P.; Gaal, K.K.; Chang, K.L.; Weiss, L.M. PDGFRA rearrangement leading to hyper-eosinophilia, T-lymphoblastic lymphoma, myeloproliferative neoplasm and precursor B-cell acute lymphoblastic leukemia. Leukemia 2011, 25, 371–375. [Google Scholar] [CrossRef]
- Trempat, P.; Villalva, C.; Laurent, G.; Armstrong, F.; Delsol, G.; Dastugue, N.; Brousset, P. Chronic myeloproliferative disorders with rearrangement of the platelet-derived growth factor alpha receptor: A new clinical target for STI571/Glivec. Oncogene 2003, 22, 5702–5706. [Google Scholar] [CrossRef]
- Akiely, R.; Almasri, F.; Almasri, N.; Abu-Ghosh, A. Case Report: Pediatric myeloid/lymphoid neoplasm with eosinophilia and PDGFRA rearrangement: The first case presenting as B-lymphoblastic lymphoma. Front. Pediatr. 2022, 10, 1059527. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef]
- Krigstein, M.; Menzies, A.; Fay, K.; Lukeis, R.; Cheung, K.; Parker, A. FIP1L1::PDGFRA fusion driving three synchronous haematological malignancies. Pathology 2023, 55, 1040–1044. [Google Scholar] [CrossRef]
- Bellani, V.; Croci, G.A.; Bucelli, C.; Maronese, C.A.; Alberti, S.; Iurlo, A.; Cattaneo, D. Lymphomatoid papulosis associated with myeloid neoplasm with eosinophilia and FIP1L1::PDGFRA rearrangement: Successful imatinib treatment in two cases. J. Dermatol. 2023, 50, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Cilloni, D.; Rondoni, M.; Ottaviani, E.; Messa, F.; Merante, S.; Tiribelli, M.; Buccisano, F.; Testoni, N.; Gottardi, E.; et al. The efficacy of imatinib mesylate in patients with FIP1L1-PDGFRalpha-positive hypereosinophilic syndrome. Results of a multicenter prospective study. Haematologica 2007, 92, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; D’Souza, A.; Knudson, R.A.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Long-term follow-up of FIP1L1-PDGFRA-mutated patients with eosinophilia: Survival and clinical outcome. Leukemia 2012, 26, 2439–2441. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Naumann, N.; Schwaab, J.; Baurmann, H.; Casper, J.; Dang, T.A.; Dietze, L.; Döhner, K.; Hänel, A.; Lathan, B.; et al. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann. Hematol. 2017, 96, 1463–1470. [Google Scholar] [CrossRef]
- Cheah, C.Y.; Burbury, K.; Apperley, J.F.; Huguet, F.; Pitini, V.; Gardembas, M.; Ross, D.M.; Forrest, D.; Genet, P.; Rousselot, P.; et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood 2014, 123, 3574–3577. [Google Scholar] [CrossRef]
- Di Giacomo, D.; Quintini, M.; Pierini, V.; Pellanera, F.; La Starza, R.; Gorello, P.; Matteucci, C.; Crescenzi, B.; Fiumara, P.F.; Veltroni, M.; et al. Genomic and clinical findings in myeloid neoplasms with PDGFRB rearrangement. Ann. Hematol. 2022, 101, 297–307. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Brunetti, M.; Stoltenberg, M.; Strandabø, R.A.U.; Staurseth, J.; Andersen, K.; Kostolomov, I.; Hveem, T.S.; Lorenz, S.; Nystad, T.A.; et al. Novel GTF2I-PDGFRB and IKZF1-TYW1 fusions in pediatric leukemia with normal karyotype. Exp. Hematol. Oncol. 2019, 8, 12. [Google Scholar] [CrossRef]
- Bain, B.J.; Fletcher, S.H. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol. Allergy Clin. N. Am. 2007, 27, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Ryan, S.L.; Chilton, L.; Elliott, A.; Murray, J.; Richardson, S.; Wragg, C.; Moppett, J.; Cummins, M.; Tunstall, O.; et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): Genetic profile and clinical implications. Blood 2016, 127, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, A.M.; Schrock, A.B.; He, J.; Nahas, M.; Curran, K.; Shukla, N.; Cramer, S.; Draper, L.; Verma, A.; Erlich, R.; et al. Novel PDGFRB Fusions in Childhood B- and T-Acute Lymphoblastic Leukemia. Leukemia 2017, 31, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, M.; Pierini, V.; Di Giacomo, D.; Zucchini, P.; Forghieri, F.; Bonacorsi, G.; Paolini, A.; Quadrelli, C.; Giacobbi, F.; Fontana, F.; et al. The Importance of Cytogenetic and Molecular Analyses in Eosinophilia-Associated Myeloproliferative Neoplasms: An Unusual Case with Normal Karyotype and TNIP1-PDGFRB Rearrangement and Overview of PDGFRB Partner Genes. Leuk. Lymphoma 2017, 58, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; Gotlib, J. Myeloid Neoplasms with Eosinophilia. Blood 2017, 129, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Darbyshire, P.J.; Shortland, D.; Swansbury, G.J.; Sadler, J.; Lawler, S.D.; Chessells, J.M. A Myeloproliferative Disease in Two Infants Associated with Eosinophilia and Chromosome t(1;5) Translocation. Br. J. Haematol. 1987, 66, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.; Velloso, E.R.; Lopes, L.F.; Lee, C.; Aster, J.C.; Shipp, M.A.; Aguiar, R.C. Cloning of the t(1;5)(q23;q33) in a Myeloproliferative Disorder Associated with Eosinophilia: Involvement of PDGFRB and Response to Imatinib. Blood 2003, 102, 4187–4190. [Google Scholar] [CrossRef]
- Li, Z.; Yang, R.; Zhao, J.; Yuan, R.; Lu, Q.; Li, Q.; Tse, W. Molecular Diagnosis and Targeted Therapy of a Pediatric Chronic Eosinophilic Leukemia Patient Carrying TPM3-PDGFRB Fusion. Pediatr. Blood Cancer 2011, 56, 463–466. [Google Scholar] [CrossRef]
- Abraham, S.; Salama, M.; Hancock, J.; Jacobsen, J.; Fluchel, M. Congenital and Childhood Myeloproliferative Disorders with Eosinophilia Responsive to Imatinib. Pediatr. Blood Cancer 2012, 59, 928–929. [Google Scholar] [CrossRef]
- Berking, A.C.; Flaadt, T.; Behrens, Y.L.; Yoshimi, A.; Leipold, A.; Holzer, U.; Lang, P.; Quintanilla-Martinez, L.; Schlegelberger, B.; Reiter, A.; et al. Rare and Potentially Fatal—Cytogenetically Cryptic TNIP1::PDGFRB and PCM1::FGFR1 Fusion Leading to Myeloid/Lymphoid Neoplasms with Eosinophilia in Children. Cancer Genet. 2023, 272–273, 29–34. [Google Scholar] [CrossRef]
- Wang, S.C.; Yang, W.Y. Myeloid Neoplasm with Eosinophilia and Rearrangement of Platelet-Derived Growth Factor Receptor Beta Gene in Children: Two Case Reports. World J. Clin. Cases 2021, 9, 204–210. [Google Scholar] [CrossRef] [PubMed]
- McKeague, S.J.; O’Rourke, K.; Fanning, S.; Joy, C.; Throp, D.; Adams, R.; Harvey, Y.; Keng, T.B. Acute Leukemia with Cytogenetically Cryptic FGFR1 Rearrangement and Lineage Switch during Therapy: A Case Report and Literature Review. Am. J. Clin. Pathol. 2023. Epub ahead of print. [Google Scholar] [CrossRef]
- Strati, P.; Tang, G.; Duose, D.Y.; Mallampati, S.; Luthra, R.; Patel, K.P.; Hussaini, M.; Mirza, A.S.; Komrokji, R.S.; Oh, S.; et al. Myeloid/Lymphoid Neoplasms with FGFR1 Rearrangement. Leuk. Lymphoma 2018, 59, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Boluda, J.C.; Pereira, A.; Zinger, N.; Gras, L.; Martino, R.; Nikolousis, E.; Finke, J.; Chinea, A.; Rambaldi, A.; Robin, M.; et al. Allogeneic Hematopoietic Cell Transplantation in Patients with Myeloid/Lymphoid Neoplasm with FGFR1-Rearrangement: A Study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. 2022, 57, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, S.; Teschemaker, A.; Kish, J.K.; Savill, K.M.; Colucci, P. Myeloid/Lymphoid Neoplasms (MLNs) with Fibroblast Growth Factor Receptor 1 (FGFR1) Rearrangement (MLNFGFR1): A US Real-World Retrospective Cohort Study. Blood 2022, 140 (Suppl. S1), 6854–6855. [Google Scholar] [CrossRef]
- Wakim, J.J.; Tirado, C.A.; Chen, W.; Collins, R. t(8;22)/BCR-FGFR1 Myeloproliferative Disorder Presenting as B-Acute Lymphoblastic Leukemia: Report of a Case Treated with Sorafenib and Review of the Literature. Leuk. Res. 2011, 35, e151–e153. [Google Scholar] [CrossRef] [PubMed]
- Haslam, K.; Langabeer, S.E.; Kelly, J.; Coen, N.; O’Connell, N.M.; Conneally, E. Allogeneic Hematopoietic Stem Cell Transplantation for a BCR-FGFR1 Myeloproliferative Neoplasm Presenting as Acute Lymphoblastic Leukemia. Case Rep. Hematol. 2012, 2012, 620967. [Google Scholar] [CrossRef] [PubMed]
- Baldazzi, C.; Iacobucci, I.; Luatti, S.; Ottaviani, E.; Marzocchi, G.; Paolini, S.; Stacchini, M.; Papayannidis, C.; Gamberini, C.; Martinelli, G.; et al. B-Cell Acute Lymphoblastic Leukemia as Evolution of a 8p11 Myeloproliferative Syndrome with t(8;22)(p11;q11) and BCR-FGFR1 Fusion Gene. Leuk. Res. 2010, 34, e282–e285. [Google Scholar] [CrossRef]
- Macdonald, D.; Aguiar, R.C.; Mason, P.J.; Goldman, J.M.; Cross, N.C. A New Myeloproliferative Disorder Associated with Chromosomal Translocations Involving 8p11: A Review. Leukemia 1995, 9, 1628–1630. [Google Scholar]
- Macdonald, D.; Reiter, A.; Cross, N.C. The 8p11 Myeloproliferative Syndrome: A Distinct Clinical Entity Caused by Constitutive Activation of FGFR1. Acta Haematol. 2002, 107, 101–107. [Google Scholar] [CrossRef]
- Nakayama, H.; Inamitsu, T.; Ohga, S.; Kai, T.; Suda, M.; Matsuzaki, A.; Ueda, K. Chronic Myelomonocytic Leukaemia with t(8;9)(p11;q34) in Childhood: An Example of the 8p11 Myeloproliferative Disorder? Br. J. Haematol. 1996, 92, 692–695. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, H.; Kroes, W.; van der Schoot, C.E.; Dee, R.; Pals, S.T.; Bouts, T.H.; Slater, R.M. A Young Child with Acquired t(8;9)(p11;q34): Additional Proof That 8p11 Is Involved in Mixed Myeloid/T Lymphoid Malignancies. Leukemia 1996, 10, 1252–1253. [Google Scholar] [PubMed]
- Wong, W.S.; Cheng, K.C.; Lau, K.M.; Chan, N.P.; Shing, M.M.; Cheng, S.H.; Chik, K.W.; Li, C.K.; Ng, M.H. Clonal Evolution of 8p11 Stem Cell Syndrome in a 14-Year-Old Chinese Boy: A Review of Literature of t(8;13) Associated Myeloproliferative Diseases. Leuk. Res. 2007, 31, 235–238. [Google Scholar] [CrossRef]
- Zhang, W.W.; Habeebu, S.; Sheehan, A.M.; Naeem, R.; Hernandez, V.S.; Dreyer, Z.E.; López-Terrada, D. Molecular Monitoring of 8p11 Myeloproliferative Syndrome in an Infant. J. Pediatr. Hematol. Oncol. 2009, 31, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Qu, S.; Liu, J.; Li, C.; Yan, X.; Xu, Z.; Qin, T.; Jia, Y.; Pan, L.; Gao, Q.; et al. Olverembatinib for Myeloid/Lymphoid Neoplasm Associated with Eosinophilia and FGFR1 Rearrangement. Leuk. Lymphoma 2023, 64, 1605–1610. [Google Scholar] [CrossRef]
- Ren, M.; Qin, H.; Ren, R.; Cowell, J.K. Ponatinib Suppresses the Development of Myeloid and Lymphoid Malignancies Associated with FGFR1 Abnormalities. Leukemia 2013, 27, 32–40. [Google Scholar] [CrossRef]
- Chen, J.; DeAngelo, D.J.; Kutok, J.L.; Williams, I.R.; Lee, B.H.; Wadleigh, M.; Duclos, N.; Cohen, S.; Adelsperger, J.; Okabe, R.; et al. PKC412 Inhibits the Zinc Finger 198-Fibroblast Growth Factor Receptor 1 Fusion Tyrosine Kinase and Is Active in Treatment of Stem Cell Myeloproliferative Disorder. Proc. Natl. Acad. Sci. USA 2004, 101, 14479–14484. [Google Scholar] [CrossRef]
- Kasbekar, M.; Nardi, V.; Dal Cin, P.; Brunner, A.M.; Burke, M.; Chen, Y.B.; Connolly, C.; Fathi, A.T.; Foster, J.; Macrae, M.; et al. Targeted FGFR Inhibition Results in a Durable Remission in an FGFR1-Driven Myeloid Neoplasm with Eosinophilia. Blood Adv. 2020, 4, 3136–3140. [Google Scholar] [CrossRef]
- Verstovsek, S.; Vannucchi, A.M.; Rambaldi, A.; Gotlib, M.J.R.; Mead, A.J.; Hochhaus, A.; Kiladjian, J.-J.; Boluda, J.C.H.; Asatiani, E.; Lihou, B.C.; et al. Interim Results from Fight-203, a Phase 2, Open-Label, Multicenter Study Evaluating the Efficacy and Safety of Pemigatinib (INCB054828) in Patients with Myeloid/Lymphoid Neoplasms with Rearrangement of Fibroblast Growth Factor Receptor 1 (FGFR1). Blood 2018, 132 (Suppl. S1), 690. [Google Scholar] [CrossRef]
- Tzankov, A.; Reichard, K.K.; Hasserjian, R.P.; Arber, D.A.; Orazi, A.; Wang, S.A. Updates on Eosinophilic Disorders. Virchows Arch. 2023, 482, 85–97. [Google Scholar] [CrossRef]
- Kaplan, H.G.; Jin, R.; Bifulco, C.B.; Scanlan, J.M.; Corwin, D.R. PCM1-JAK2 Fusion Tyrosine Kinase Gene-Related Neoplasia: A Systematic Review of the Clinical Literature. Oncologist 2022, 27, e661–e670. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Milosevic, J.D.; Casetti, I.; Dambruoso, I.; Pietra, D.; Boveri, E.; Boni, M.; Bernasconi, P.; Passamonti, F.; Kralovics, R.; et al. Efficacy of Ruxolitinib in Chronic Eosinophilic Leukemia Associated with a PCM1-JAK2 Fusion Gene. J. Clin. Oncol. 2013, 31, e269–e271. [Google Scholar] [CrossRef] [PubMed]
- Lierman, E.; Selleslag, D.; Smits, S.; Billiet, J.; Vandenberghe, P. Ruxolitinib Inhibits Transforming JAK2 Fusion Proteins In Vitro and Induces Complete Cytogenetic Remission in t(8;9)(p22;p24)/PCM1-JAK2-Positive Chronic Eosinophilic Leukemia. Blood 2012, 120, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Tam, W.; Short, N.J.; Bose, P.; Wu, D.; Hurwitz, S.N.; Bagg, A.; Rogers, H.J.; Hsi, E.D.; Quesada, A.E.; et al. Myeloid/Lymphoid Neoplasms with FLT3 Rearrangement. Mod. Pathol. 2021, 34, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Venable, E.R.; Gagnon, M.F.; Pitel, B.A.; Palmer, J.M.; Peterson, J.F.; Baughn, L.B.; Hoppman, N.L.; Greipp, P.T.; Ketterling, R.P.; Patnaik, M.S.; et al. A TRIP11::FLT3 Gene Fusion in a Patient with Myeloid/Lymphoid Neoplasm with Eosinophilia and Tyrosine Kinase Gene Fusions: A Case Report and Review of the Literature. Cold Spring Harb. Mol. Case Stud. 2023, 9, a006243. [Google Scholar] [CrossRef]
- Schoelinck, J.; Gervasoni, J.; Guillermin, Y.; Beillard, E.; Pissaloux, D.; Chassagne-Clement, C. T Cell Phenotype and Lack of Eosinophilia Are Not Uncommon in Extramedullary Myeloid/Lymphoid Neoplasms with ETV6::FLT3 Fusion: A Case Report and Review of the Literature. Virchows Arch. 2023. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, B.; Dela Cruz, F.S.; Ibanez Sanchez, G.D.; Zhang, Y.; Xiao, W.; Benayed, R.; Markova, A.; Rodriguez-Sanchez, M.I.; Bouvier, N.; Roshal, M.; et al. ETV6-FLT3-Positive Myeloid/Lymphoid Neoplasm with Eosinophilia Presenting in an Infant: An Entity Distinct from JMML. Blood Adv. 2021, 5, 1899–1902. [Google Scholar] [CrossRef]
- Munthe-Kaas, M.C.; Forthun, R.B.; Brendehaug, A.; Eek, A.K.; Høysæter, T.; Osnes, L.T.N.; Prescott, T.; Spetalen, S.; Hovland, R. Partial Response to Sorafenib in a Child with a Myeloid/Lymphoid Neoplasm, Eosinophilia, and a ZMYM2-FLT3 Fusion. J. Pediatr. Hematol. Oncol. 2021, 43, e508–e511. [Google Scholar] [CrossRef]
- Falchi, L.; Mehrotra, M.; Newberry, K.J.; Lyle, L.M.; Lu, G.; Patel, K.P.; Luthra, R.; Popat, U.; Verstovsek, S. ETV6-FLT3 Fusion Gene-Positive, Eosinophilia-Associated Myeloproliferative Neoplasm Successfully Treated with Sorafenib and Allogeneic Stem Cell Transplant. Leukemia 2014, 28, 2090–2092. [Google Scholar] [CrossRef]
- Walz, C.; Erben, P.; Ritter, M.; Bloor, A.; Metzgeroth, G.; Telford, N.; Haferlach, C.; Haferlach, T.; Gesk, S.; Score, J.; et al. Response of ETV6-FLT3-Positive Myeloid/Lymphoid Neoplasm with Eosinophilia to Inhibitors of FMS-Like Tyrosine Kinase 3. Blood 2011, 118, 2239–2242. [Google Scholar] [CrossRef]
- Chonabayashi, K.; Hishizawa, M.; Matsui, M.; Kondo, T.; Ohno, T.; Ishikawa, T.; Takaori-Kondo, A. Successful Allogeneic Stem Cell Transplantation with Long-Term Remission of ETV6/FLT3-Positive Myeloid/Lymphoid Neoplasm with Eosinophilia. Ann. Hematol. 2014, 93, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- De Braekeleer, E.; Douet-Guilbert, N.; Rowe, D.; Bown, N.; Morel, F.; Berthou, C.; Férec, C.; De Braekeleer, M. ABL1 Fusion Genes in Hematological Malignancies: A Review. Eur. J. Haematol. 2011, 86, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Moorman, A.V.; Cazzaniga, G.; Stanulla, M.; Harvey, R.C.; Roberts, K.G.; Heatley, S.L.; Loh, M.L.; Konopleva, M.; Chen, I.M.; et al. Characterization of Leukemias with ETV6-ABL1 Fusion. Haematologica 2016, 101, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Naumann, N.; Luebke, J.; Jawhar, M.; Somervaille, T.C.P.; Williams, M.S.; Frewin, R.; Jost, P.J.; Lichtenegger, F.S.; La Rosée, P.; et al. Response to Tyrosine Kinase Inhibitors in Myeloid Neoplasms Associated with PCM1-JAK2, BCR-JAK2, and ETV6-ABL1 Fusion Genes. Am. J. Hematol. 2020, 95, 824–833. [Google Scholar] [CrossRef]
- Carll, T.; Patel, A.; Derman, B.; Hyjek, E.; Lager, A.; Wanjari, P.; Segal, J.; Odenike, O.; Fidai, S.; Arber, D. Diagnosis and Treatment of Mixed Phenotype (T-Myeloid/Lymphoid) Acute Leukemia with Novel ETV6-FGFR2 Rearrangement. Blood Adv. 2020, 4, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- Telford, N.; Alexander, S.; McGinn, O.J.; Williams, M.; Wood, K.M.; Bloor, A.; Saha, V. Myeloproliferative Neoplasm with Eosinophilia and T-Lymphoblastic Lymphoma with ETV6-LYN Gene Fusion. Blood Cancer J. 2016, 6, e412. [Google Scholar] [CrossRef] [PubMed]
- Kralik, J.M.; Kranewitter, W.; Boesmueller, H.; Marschon, R.; Tschurtschenthaler, G.; Rumpold, H.; Wiesinger, K.; Erdel, M.; Petzer, A.L.; Webersinke, G. Characterization of a Newly Identified ETV6-NTRK3 Fusion Transcript in Acute Myeloid Leukemia. Diagn. Pathol. 2011, 6, 19. [Google Scholar] [CrossRef]
- Maesako, Y.; Izumi, K.; Okamori, S.; Takeoka, K.; Kishimori, C.; Okumura, A.; Honjo, G.; Akasaka, T.; Ohno, H. Inv(2)(p23q13)/RAN-Binding Protein 2 (RANBP2)-ALK Fusion Gene in Myeloid Leukemia That Developed in an Elderly Woman. Int. J. Hematol. 2014, 99, 202–207. [Google Scholar] [CrossRef]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.F.; et al. RET Fusion Genes Are Associated with Chronic Myelomonocytic Leukemia and Enhance Monocytic Differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef]
- Roberts, K.G.; Janke, L.J.; Zhao, Y.; Seth, A.; Ma, J.; Finkelstein, D.; Smith, S.; Ebata, K.; Tuch, B.B.; Hunger, S.P.; et al. ETV6-NTRK3 Induces Aggressive Acute Lymphoblastic Leukemia Highly Sensitive to Selective TRK Inhibition. Blood 2018, 132, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Maesako, Y.; Okumura, A.; Takeoka, K.; Kishimori, C.; Izumi, K.; Kamoda, Y.; Iioka, F.; Akasaka, T.; Ohno, H. Reduction of Leukemia Cell Burden and Restoration of Normal Hematopoiesis at 3 Months of Crizotinib Treatment in RAN-Binding Protein 2 (RANBP2)-Anaplastic Lymphoma Kinase (ALK) Acute Myeloid Leukemia. Leukemia 2014, 28, 1935–1937. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Marcellino, B.; Mascarenhas, J. Eosinophilia in Acute Myeloid Leukemia: Overlooked and Underexamined. Blood Rev. 2019, 36, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Williams, S.F.; Le Beau, M.M.; Bitter, M.A.; Vardiman, J.W.; Rowley, J.D. Acute Myelomonocytic Leukemia with Abnormal Eosinophils and Inv(16) or t(16;16) Has a Favorable Prognosis. Blood 1986, 68, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Goswami, M.; Peng, J.; Zuo, Z.; Daver, N.; Borthakur, G.; Tang, G.; Medeiros, L.J.; Jorgensen, J.L.; Ravandi, F.; et al. Comparison of Multiparameter Flow Cytometry Immunophenotypic Analysis and Quantitative RT-PCR for the Detection of Minimal Residual Disease of Core Binding Factor Acute Myeloid Leukemia. Am. J. Clin. Pathol. 2016, 145, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Raya, J.M.; Gómez-Hernando, M.; Guijarro, F.; Alonso, E.; Arnan, M.; Senent, L.; Vicente, A.I.; Borrego, A.; Gómez-Casares, M.T.; González-González, C.; et al. PO-115: Leucemia Aguda Mieloide con t(8,21); RUNX1-RUNX1T1: Análisis Multicéntrico Retrospectivo de 165 Pacientes. In Proceedings of the LXIII Congreso Nacional de la SEHH XXXVII Congreso Nacional de la SETH, Pamplona, Spain, 14–16 October 2021; 2021. [Google Scholar]
- Swirsky, D.M.; Li, Y.S.; Matthews, J.G.; Flemans, R.J.; Rees, J.K.; Hayhoe, F.G. 8;21 Translocation in Acute Granulocytic Leukaemia: Cytological, Cytochemical and Clinical Features. Br. J. Haematol. 1984, 56, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Bennett, J.M.; Löffler, H.; Gassmann, W.; Andersen, J.W.; Tuzuner, N.; Casslleth, P.A.; Fonatsch, C.; Schoch, C.; Schlegelberger, B.; et al. Acute Myeloid Leukemia with Translocation (8;21). Cytomorphology, Dysplasia, and Prognostic Factors in 41 Cases. AML Cooperative Group and ECOG. Leuk. Lymphoma 1996, 23, 227–234. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Hemasphere 2021, 5, e646. [Google Scholar] [CrossRef]
- Kluin-Nelemans, H.C.; Reiter, A.; Illerhaus, A.; van Anrooij, B.; Hartmann, K.; Span, L.F.R.; Gorska, A.; Niedoszytko, M.; Lange, M.; Scaffidi, L.; et al. Prognostic Impact of Eosinophils in Mastocytosis: Analysis of 2350 Patients Collected in the ECNM Registry. Leukemia 2020, 34, 1090–1101. [Google Scholar] [CrossRef]
- Kovalszki, A.; Weller, P.F. Eosinophilia in Mast Cell Disease. Immunol. Allergy Clin. N. Am. 2014, 34, 357–364. [Google Scholar] [CrossRef]
- Morales-Camacho, R.M.; Villanueva-Herraiz, S.; Prats-Martín, C.; Borrero, J.J.; Bernal, R.; Vargas, M.T. Eosinophil Phagocytosis in Advanced Systemic Mastocytosis with Eosinophilia. Br. J. Haematol. 2018, 181, 578. [Google Scholar] [CrossRef] [PubMed]
- Moshref Razavi, H.; Yu, R. Myeloid Neoplasm with PDGFRB Rearrangement, Presenting as Systemic Mastocytosis-Chronic Eosinophilic Leukemia. Am. J. Hematol. 2022, 97, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Leguit, R.J.; Wang, S.A.; George, T.I.; Tzankov, A.; Orazi, A. The International Consensus Classification of Mastocytosis and Related Entities. Virchows Arch. 2023, 482, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Morgado, J.M.; Perbellini, O.; Johnson, R.C.; Teodósio, C.; Matito, A.; Álvarez-Twose, I.; Bonadonna, P.; Zamò, A.; Jara-Acevedo, M.; Mayado, A.; et al. CD30 Expression by Bone Marrow Mast Cells from Different Diagnostic Variants of Systemic Mastocytosis. Histopathology 2013, 63, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, C.B.; Zhang, L.; Li, S. Systemic Mastocytosis with Associated Myeloproliferative Neoplasm with t(8;19)(p12;q13.1) and Abnormality of FGFR1: Report of a Unique Case. Int. J. Clin. Exp. Pathol. 2014, 7, 801–807. [Google Scholar] [PubMed]
- Langabeer, S.E. The Eosinophilic Variant of Chronic Myeloid Leukemia. EXCLI J. 2021, 20, 1608–1609. [Google Scholar] [CrossRef] [PubMed]
- Morsia, E.; Reichard, K.; Pardanani, A.; Tefferi, A.; Gangat, N. WHO Defined Chronic Eosinophilic Leukemia, Not Otherwise Specified (CEL, NOS): A Contemporary Series from the Mayo Clinic. Am. J. Hematol. 2020, 95, E172–E174. [Google Scholar] [CrossRef]
- Matsushima, T.; Handa, H.; Yokohama, A.; Nagasaki, J.; Koiso, H.; Kin, Y.; Tanaka, Y.; Sakura, T.; Tsukamoto, N.; Karasawa, M.; et al. Prevalence and Clinical Characteristics of Myelodysplastic Syndrome with Bone Marrow Eosinophilia or Basophilia. Blood 2003, 101, 3386–3390. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Gangat, N.; Knudson, R.A.; Keefe, J.G.; Hanson, C.A.; Pardanani, A.; Ketterling, R.P.; Tefferi, A. Chromosome 8p11.2 Translocations: Prevalence, FISH Analysis for FGFR1 and MYST3, and Clinicopathologic Correlates in a Consecutive Cohort of 13 Cases from a Single Institution. Am. J. Hematol. 2010, 85, 238–242. [Google Scholar] [CrossRef]
- Tang, G.; Sydney Sir Philip, J.K.; Weinberg, O.; Tam, W.; Sadigh, S.; Lake, J.I.; Margolskee, E.M.; Rogers, H.J.; Miranda, R.N.; Bueso-Ramos, C.C.; et al. Hematopoietic Neoplasms with 9p24/JAK2 Rearrangement: A Multicenter Study. Mod. Pathol. 2019, 32, 490–498. [Google Scholar] [CrossRef]













| Eosinophilia Categories | Supportive Features | ||
|---|---|---|---|
| Familial (hereditary) eosinophilia (with/without immunodeficiency) | Age, family history, and genetic study | ||
| Secondary (reactive) eosinophilia | Non-hematological |
| Clinical history and work-up focused on secondary causes |
| Hematological | Lymphoid diseases | Hematologic work-up * | |
| Primary (clonal, neoplastic) eosinophilia | Stem cell and myeloid neoplasms | Hematologic work-up * | |
| Eosinophilia of unknown significance | Exclusion of previous disorders or conditions | ||
Hematological neoplasms associated with reactive or secondary eosinophilia *
|
Hematological neoplasm associated with neoplastic or primary eosinophilia **
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Camacho, R.M.; Caballero-Velázquez, T.; Borrero, J.J.; Bernal, R.; Prats-Martín, C. Hematological Neoplasms with Eosinophilia. Cancers 2024, 16, 337. https://doi.org/10.3390/cancers16020337
Morales-Camacho RM, Caballero-Velázquez T, Borrero JJ, Bernal R, Prats-Martín C. Hematological Neoplasms with Eosinophilia. Cancers. 2024; 16(2):337. https://doi.org/10.3390/cancers16020337
Chicago/Turabian StyleMorales-Camacho, Rosario M., Teresa Caballero-Velázquez, Juan José Borrero, Ricardo Bernal, and Concepción Prats-Martín. 2024. "Hematological Neoplasms with Eosinophilia" Cancers 16, no. 2: 337. https://doi.org/10.3390/cancers16020337
APA StyleMorales-Camacho, R. M., Caballero-Velázquez, T., Borrero, J. J., Bernal, R., & Prats-Martín, C. (2024). Hematological Neoplasms with Eosinophilia. Cancers, 16(2), 337. https://doi.org/10.3390/cancers16020337