
Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts

 , , , ,
, , , ,  , add
Show full author list
, add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo Treatment
2.1.1. Mouse–Human F1/F2 PDX Creation/Tumor Engraftment
2.1.2. Opaganib and Upamostat Treatment of PDX Tumor-Bearing Mice
2.1.3. Pharmacokinetic (PK) Analysis
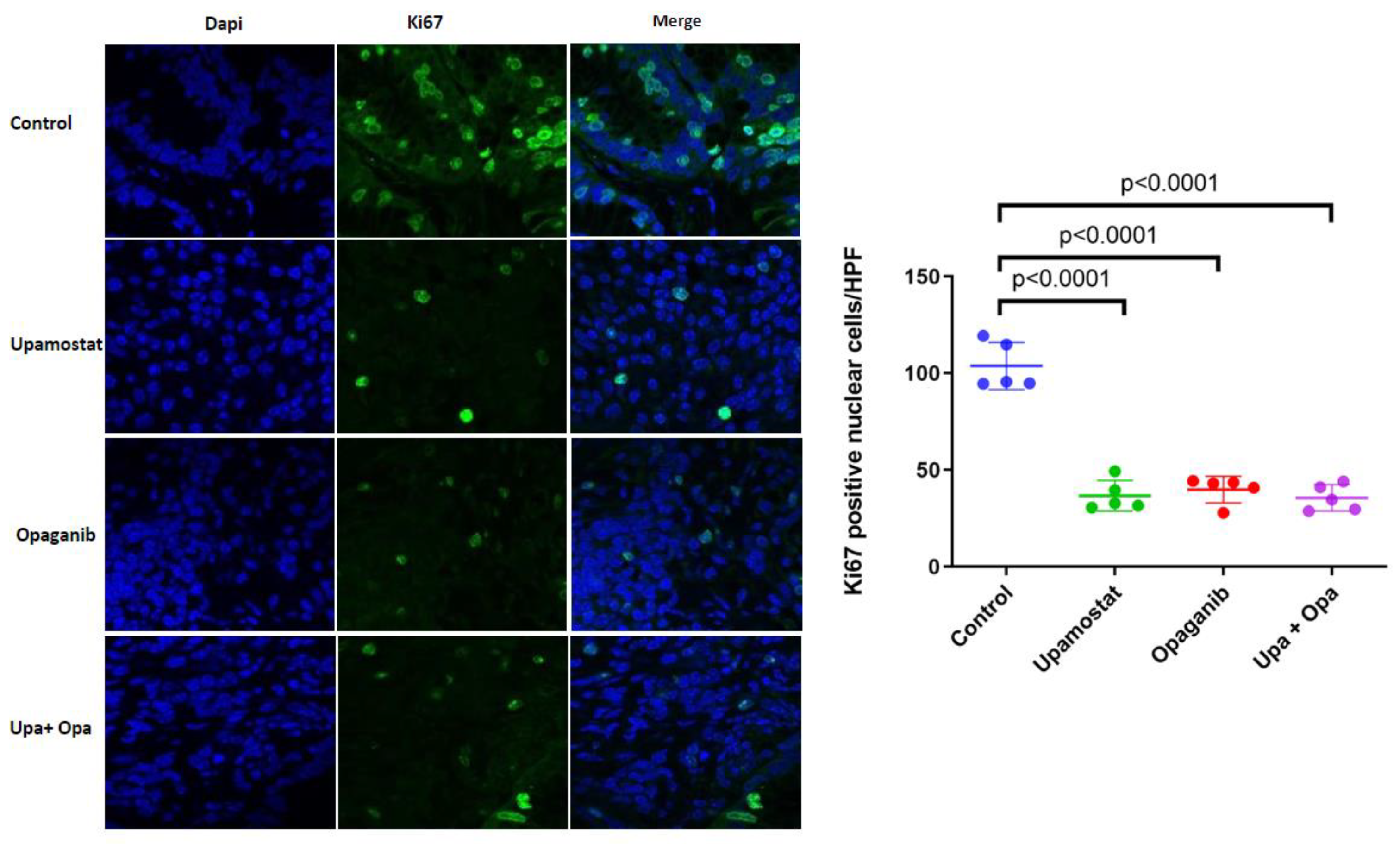
2.1.4. Ki67 Cell Proliferation Analysis
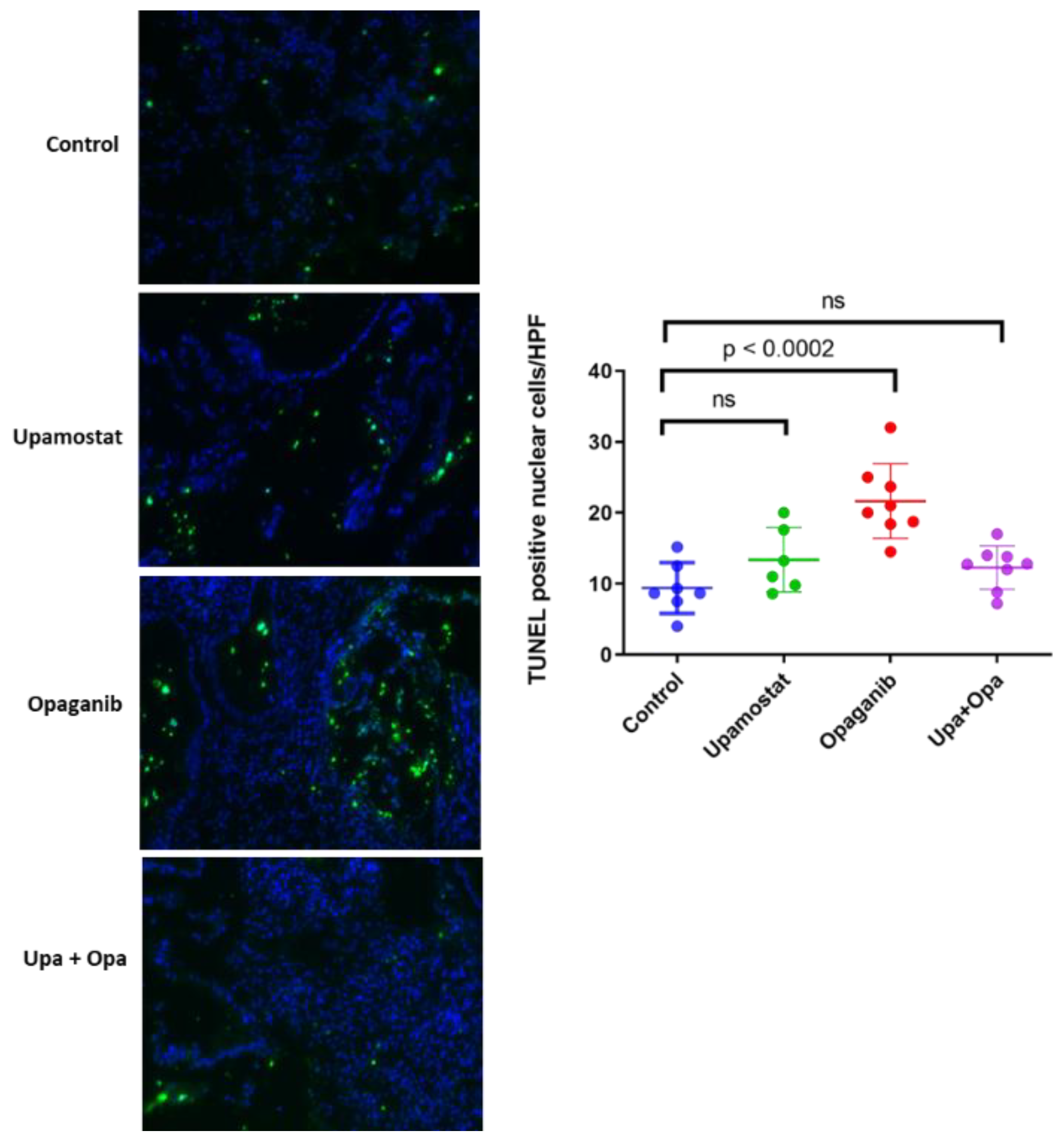
2.1.5. TUNEL Assay for Cell Death Detection
2.1.6. Immunohistochemistry
2.2. In Vitro Assay and Staining
2.2.1. Cholangiocarcinoma Cell Line
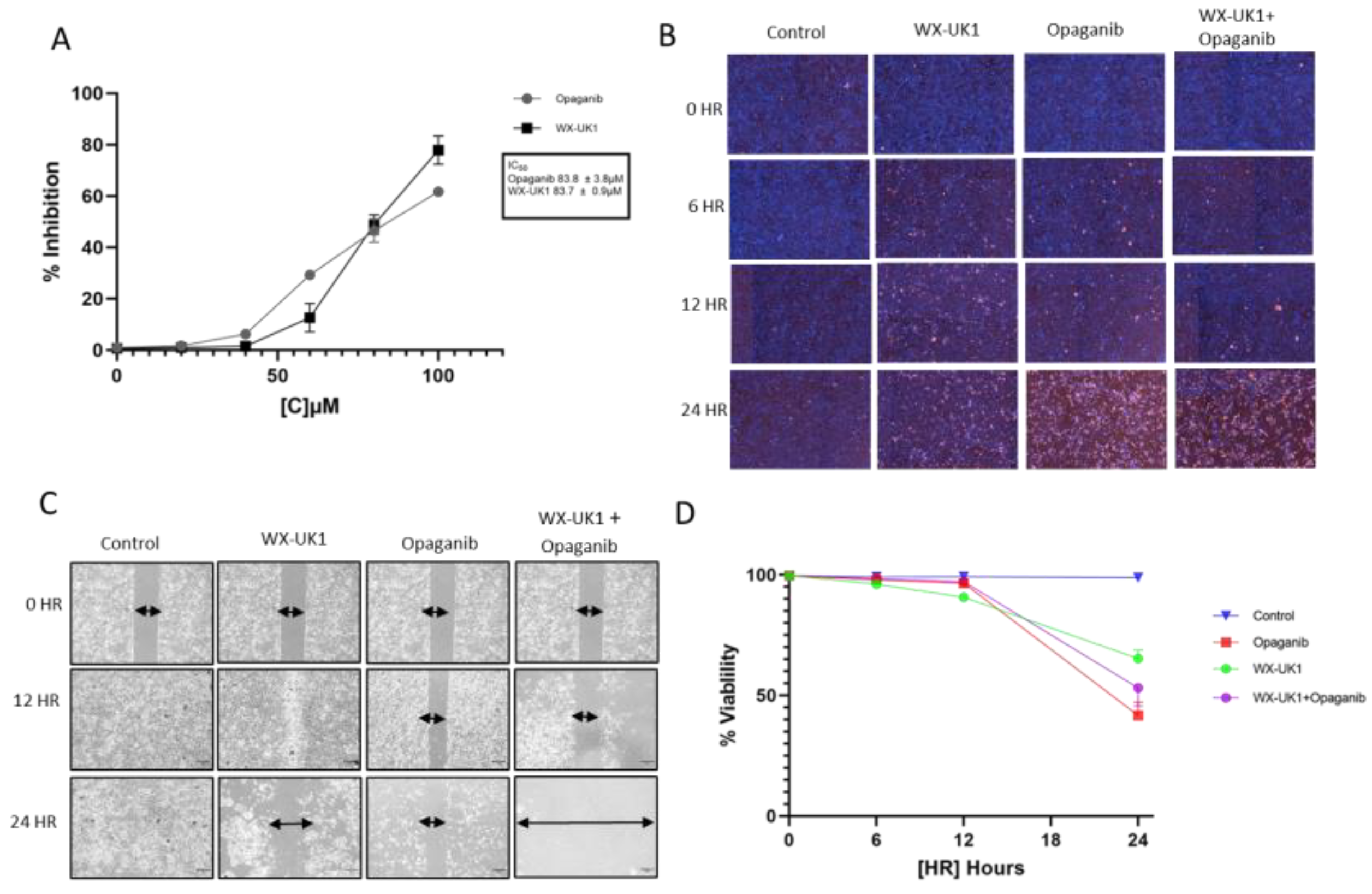
2.2.2. Cell Migration Assay
2.2.3. Cell Viability Assay
2.3. Data and Statistical Analysis
3. Results
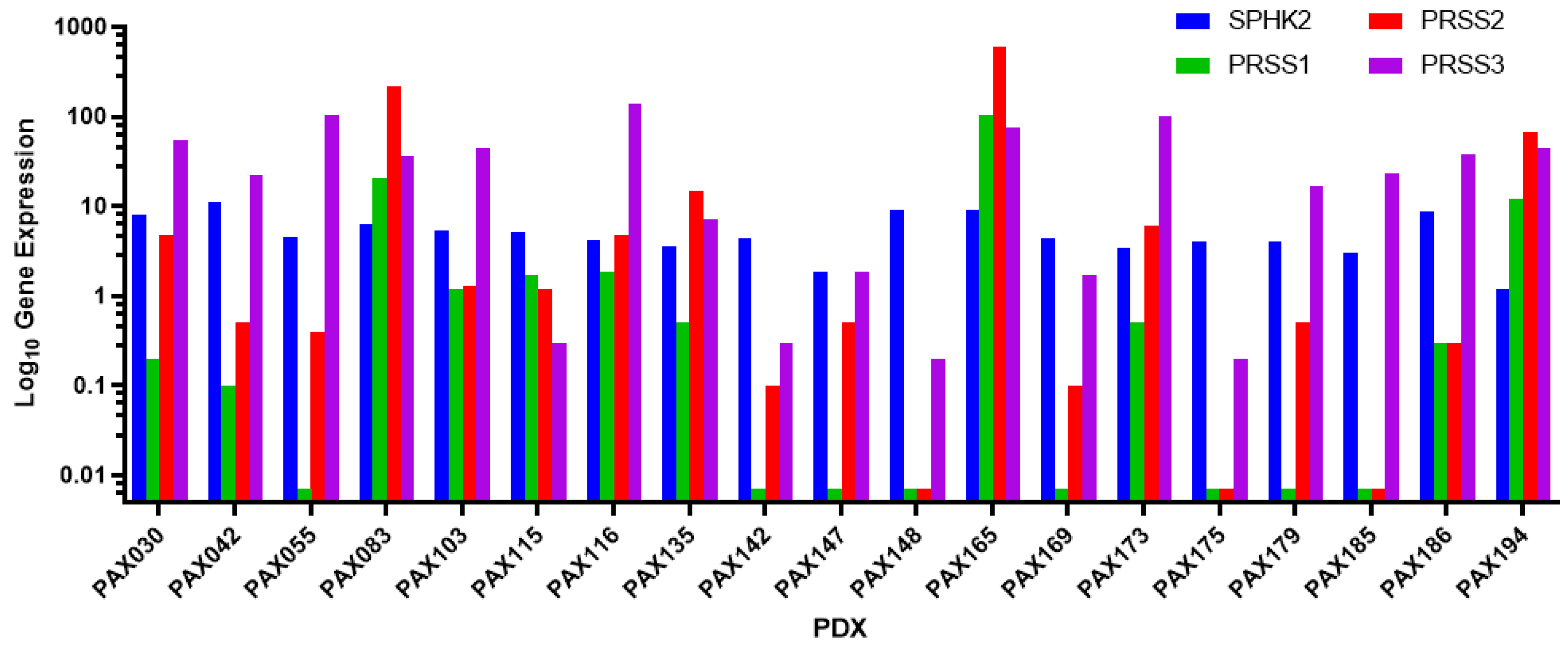
3.1. PAX165 Expresses Significant Levels of Sphingosine Kinase 2 and Serine Proteases 1, 2 and 3
3.2. Verification of Likeness between Original Tumor and Xenografts
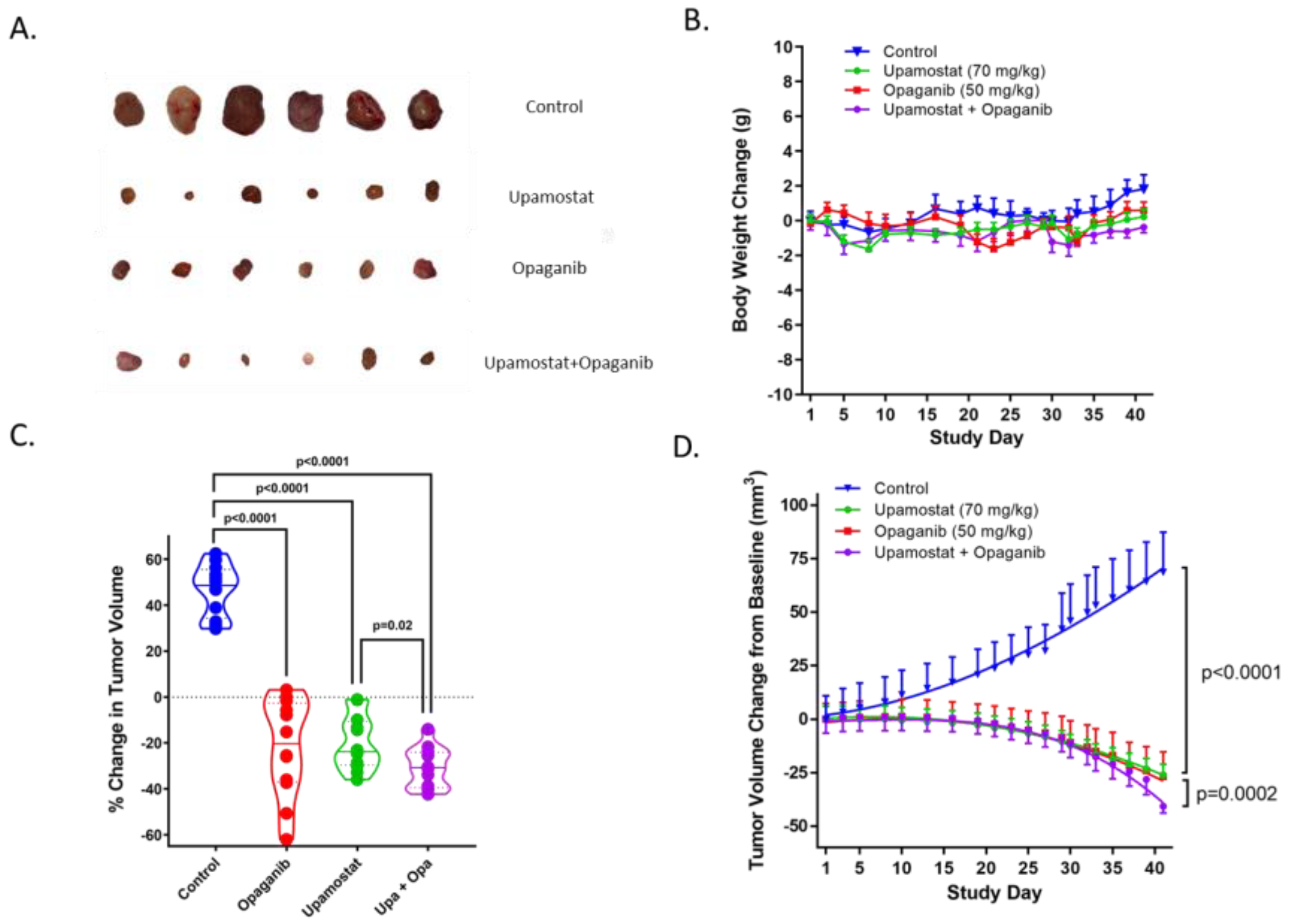
3.3. Opaganib and Upamostat Suppress CCA Tumor Growth in Mice
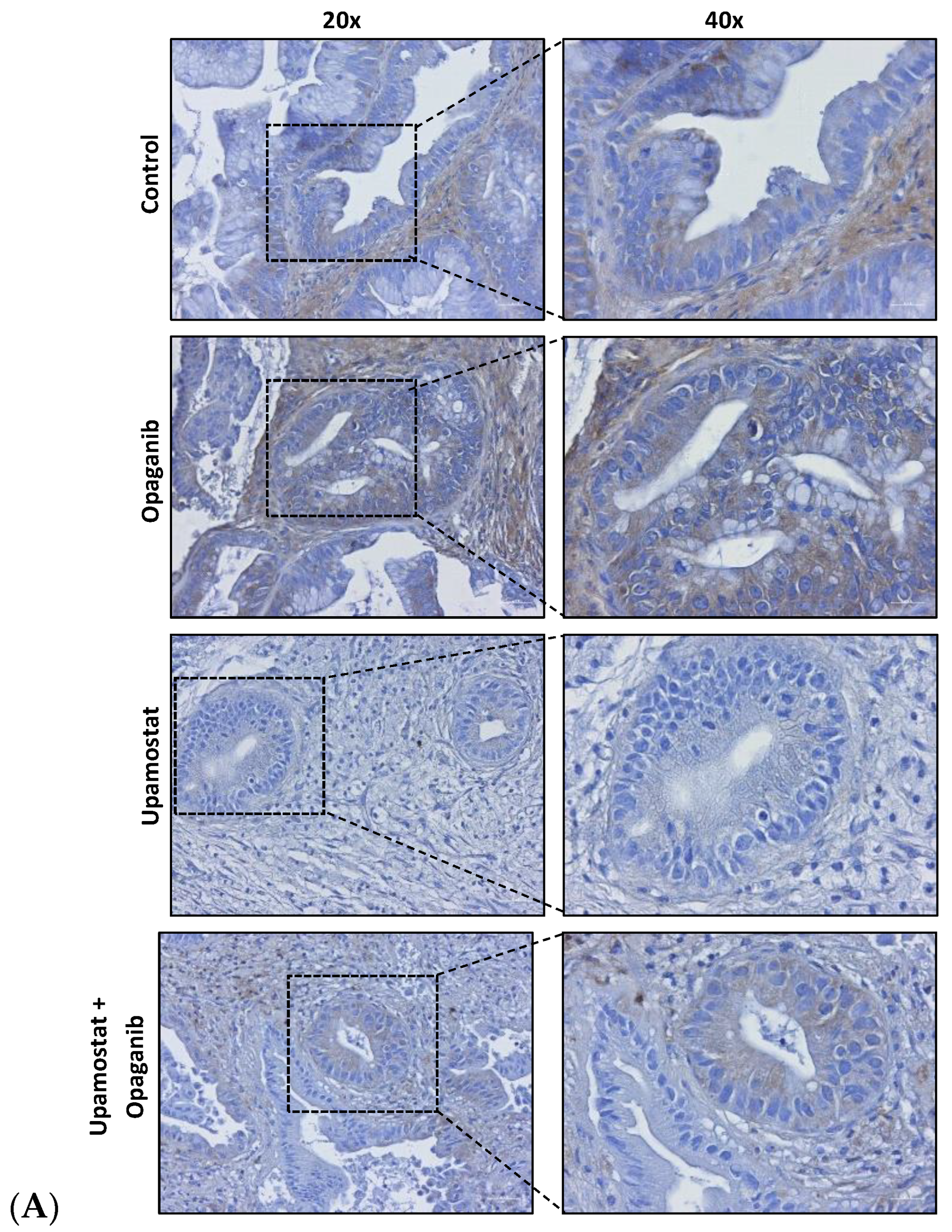
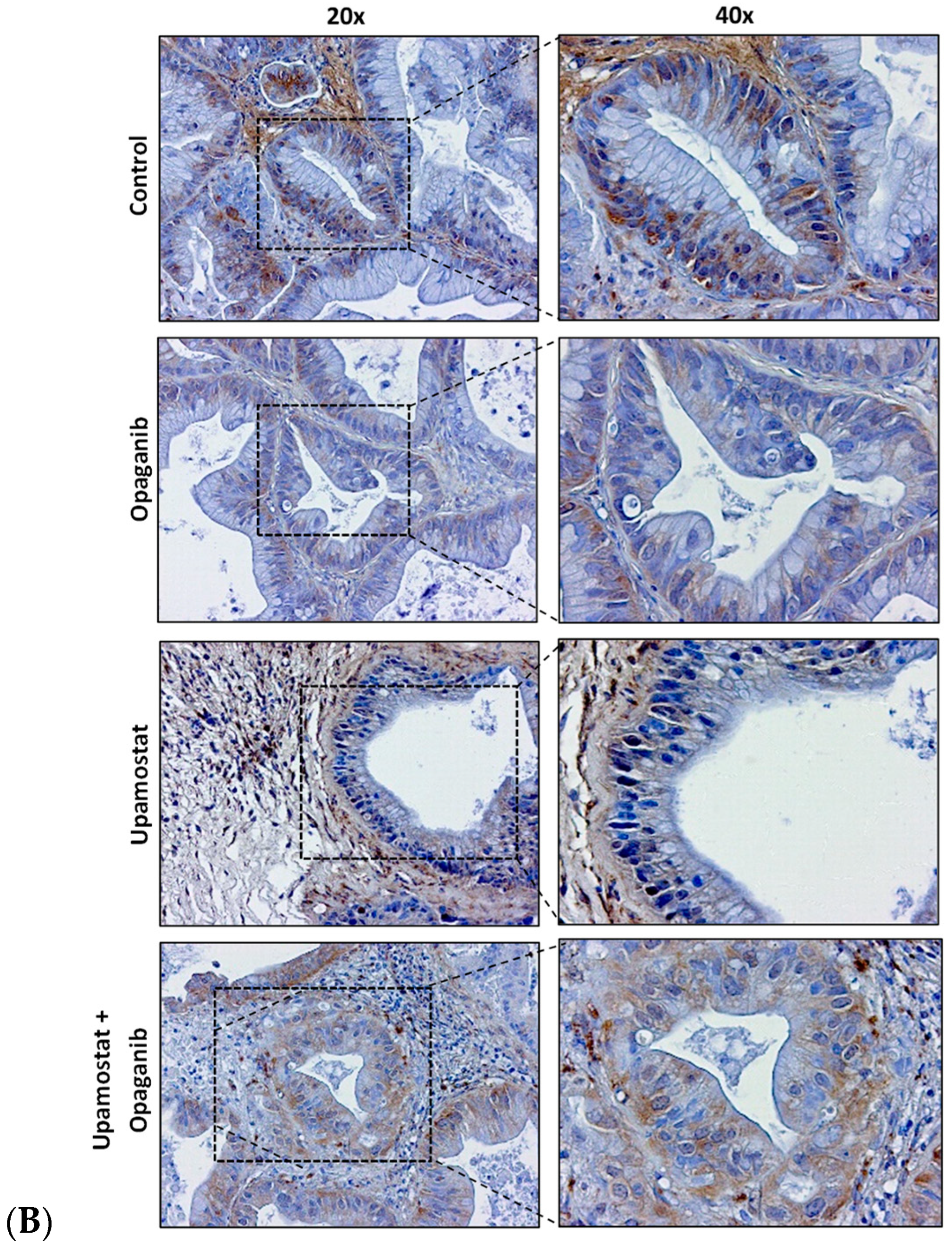
3.4. Opaganib and Upamostat Modulate Drug Target Expression in CCA Tumors
3.5. Opaganib and Upamostat Inhibit Proliferation and Induce Apoptosis in CCA Tumors
3.6. Pharmacokinetic (PK) Analysis Confirmed That Upamostat Is Metabolized to WX-UK1
3.7. Cell Viability and Migration in CCA Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. Available online: https://acsjournals.onlinelibrary.wiley.com/doi/epdf/10.3322/caac.21660 (accessed on 24 April 2023). [CrossRef] [PubMed]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, A. Clinical Presentation, Diagnosis and Staging of Cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, Z.; Guo, P.; Wang, Y.; Chen, G. Therapy for Advanced Cholangiocarcinoma: Current Knowledge and Future Potential. J. Cell Mol. Med. 2021, 25, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. REVIEW|New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Czauderna, C.; Kirstein, M.M.; Tews, H.C.; Vogel, A.; Marquardt, J.U. Molecular Subtypes and Precision Oncology in Intrahepatic Cholangiocarcinoma. J. Clin. Med. 2021, 10, 2803. [Google Scholar] [CrossRef]
- Oldenburg, E.; Schar, C.R.; Lange, E.; Plasse, T.F.; Abramson, D.T.; Fathi, R.; Towler, E.M.; Levitt, M.; Jensen, J.K. Abstract 4200: New Potential Therapeutic Applications of WX-UK1, as a Specific and Potent Inhibitor of Human Trypsin-like Proteases. Cancer Res. 2018, 78, 4200. [Google Scholar] [CrossRef]
- Setyono-Han, B.; Stürzebecher, J.; Schmalix, W.A.; Muehlenweg, B.; Sieuwerts, A.M.; Timmermans, M.; Magdolen, V.; Schmitt, M.; Klijn, J.G.M.; Foekens, J.A. Suppression of Rat Breast Cancer Metastasis and Reduction of Primary Tumour Growth by the Small Synthetic Urokinase Inhibitor WX-UK1. Thromb. Haemost. 2005, 93, 779–786. [Google Scholar] [CrossRef]
- Ertongur, S.; Lang, S.; Mack, B.; Wosikowski, K.; Muehlenweg, B.; Gires, O. Inhibition of the Invasion Capacity of Carcinoma Cells by WX-UK1, a Novel Synthetic Inhibitor of the Urokinase-Type Plasminogen Activator System. Int. J. Cancer 2004, 110, 815–824. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular Matrix (ECM) Stiffness and Degradation as Cancer Drivers. J. Cell. Biochem. 2019, 120, 2782–2790. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcb.27681 (accessed on 16 March 2023). [CrossRef]
- Hildenbrand, R.; Gandhari, M.; Stroebel, P.; Marx, A.; Allgayer, H.; Arens, N. The Urokinase-System--Role of Cell Proliferation and Apoptosis. Histol. Histopathol. 2008, 23, 227–236. [Google Scholar] [CrossRef]
- Mazar, A.P. Urokinase Plasminogen Activator Receptor Choreographs Multiple Ligand Interactions: Implications for Tumor Progression and Therapy. Clin. Cancer Res. 2008, 14, 5649–5655. [Google Scholar] [CrossRef] [PubMed]
- Sidenius, N.; Blasi, F. The Urokinase Plasminogen Activator System in Cancer: Recent Advances and Implication for Prognosis and Therapy. Cancer Metastasis Rev. 2003, 22, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.R.; Mihaly, J.; Prager, G.W. UPAR-UPA-PAI-1 Interactions and Signaling: A Vascular Biologist’s View. Thromb. Haemost. 2007, 97, 336–342. [Google Scholar]
- Bauer, T.W.; Fan, F.; Liu, W.; Johnson, M.; Parikh, N.U.; Parry, G.C.; Callahan, J.; Mazar, A.P.; Gallick, G.E.; Ellis, L.M. Insulinlike Growth Factor-I-Mediated Migration and Invasion of Human Colon Carcinoma Cells Requires Activation of c-Met and Urokinase Plasminogen Activator Receptor. Ann. Surg. 2005, 241, 748–758. [Google Scholar] [CrossRef]
- Rabbani, S.; Harakidas, P.; Davidson, D.J.; Henkin, J.; Mazar, A.P. Prevention of Prostate-Cancer Metastasis in Vivo by a Novel Synthetic Inhibitor of Urokinase-Type Plasminogen Activator (UPA). Int. J. Cancer 1995, 63, 840–845. [Google Scholar] [CrossRef]
- Durand, M.K.V.; Bødker, J.S.; Christensen, A.; Dupont, D.M.; Hansen, M.; Jensen, J.K.; Kjelgaard, S.; Mathiasen, L.; Pedersen, K.E.; Skeldal, S.; et al. Plasminogen Activator Inhibitor-1 and Tumour Growth, Invasion, and Metastasis. Thromb. Haemost. 2004, 91, 438–449. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.W.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated Bile Acids Promote Cholangiocarcinoma Cell Invasive Growth through Activation of Sphingosine 1-Phosphate Receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef]
- Song, K.; Dai, L.; Long, X.; Cui, X.; Liu, Y.; Di, W. Sphingosine Kinase 2 Inhibitor ABC294640 Displays Anti-Epithelial Ovarian Cancer Activities in Vitro and in Vivo. Onco Targets Ther. 2019, 12, 4437–4449. [Google Scholar] [CrossRef]
- Xun, C.; Chen, M.-B.; Qi, L.; Tie-Ning, Z.; Peng, X.; Ning, L.; Zhi-Xiao, C.; Li-Wei, W. Targeting Sphingosine Kinase 2 (SphK2) by ABC294640 Inhibits Colorectal Cancer Cell Growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. [Google Scholar] [CrossRef] [PubMed]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of Sphingosine Kinase 2 Downregulates the Expression of C-Myc and Mcl-1 and Induces Apoptosis in Multiple Myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, Y.; Huang, T.; Xu, G.; Peng, C.; Chen, G.; Kong, B.; Friess, H.; Shen, S.; Lv, Y.; et al. Targeting Sphingosine Kinase 2 Suppresses Cell Growth and Synergizes with BCL2/BCL-XL Inhibitors through NOXA-Mediated MCL1 Degradation in Cholangiocarcinoma. Am. J. Cancer Res. 2019, 9, 546–561. [Google Scholar]
- Ding, X.; Chaiteerakij, R.; Moser, C.D.; Shaleh, H.; Boakye, J.; Chen, G.; Ndzengue, A.; Li, Y.; Zhou, Y.; Huang, S.; et al. Antitumor Effect of the Novel Sphingosine Kinase 2 Inhibitor ABC294640 Is Enhanced by Inhibition of Autophagy and by Sorafenib in Human Cholangiocarcinoma Cells. Oncotarget 2016, 7, 20080–20092. [Google Scholar] [CrossRef]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef]
- Heinemann, V.; Ebert, M.P.; Laubender, R.P.; Bevan, P.; Mala, C.; Boeck, S. Phase II Randomised Proof-of-Concept Study of the Urokinase Inhibitor Upamostat (WX-671) in Combination with Gemcitabine Compared with Gemcitabine Alone in Patients with Non-Resectable, Locally Advanced Pancreatic Cancer. Br. J. Cancer 2013, 108, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.E.; Brocks, C.; Graefe, H.; Mala, C.; Thäns, N.; Bürgle, M.; Rempel, A.; Rotter, N.; Wollenberg, B.; Lang, S. The Oral Serine Protease Inhibitor WX-671—First Experience in Patients with Advanced Head and Neck Carcinoma. Breast Care 2008, 3, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.C.; Yang, L.; Leiting, J.L.; Sugihara, T.; Bergquist, J.R.; Ivanics, T.; Graham, R.; Truty, M.J. Successful Secondary Engraftment of Pancreatic Ductal Adenocarcinoma and Cholangiocarcinoma Patient-Derived Xenografts After Previous Failed Primary Engraftment. Transl. Oncol. 2019, 12, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Ivanics, T.; Bergquist, J.R.; Liu, G.; Kim, M.P.; Kang, Y.; Katz, M.H.; Perez, M.V.R.; Thomas, R.M.; Fleming, J.B.; Truty, M.J. Patient-derived xenograft cryopreservation and reanimation outcomes are dependent on cryoprotectant type. Lab. Investig. 2018, 98, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Leiting, J.L.; Murphy, S.J.; Bergquist, J.R.; Hernandez, M.C.; Ivanics, T.; Abdelrahman, A.M.; Yang, L.; Lynch, I.; Smadbeck, J.B.; Cleary, S.P.; et al. Biliary tract cancer patient-derived xenografts: Surgeon impact on individualized medicine. JHEP Rep. Innov. Hepatol. 2020, 2, 100068. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, P.; Devaney, D.; O’Siorain, L.; Fennelly, J.J.; Lijnen, H.J. Urokinase-plasminogen activator, a marker for aggressive breast carcinomas. Preliminary report. Cancer 1988, 62, 531–533. [Google Scholar] [CrossRef]
- Reuning, U.; Sperl, S.; Kopitz, C.; Kessler, H.; Kruger, A.; Schmitt, M.; Magdolen, V. Urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Development of antagonists of uPA/uPAR interaction and their effects in vitro and in vivo. Curr. Pharm. Des. 2003, 9, 1529–1543. [Google Scholar] [CrossRef]
- Blasi, F.; Vassalli, J.D.; Danø, K. Urokinase-type plasminogen activator: Proenzyme, receptor, and inhibitors. J. Cell Biol. 1987, 104, 801–804. [Google Scholar] [CrossRef]
- Reuning, U.; Magdolen, V.; Wilhelm, O.; Fischer, K.; Lutz, V.; Graeff, H.; Schmitt, M. Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis (review). Int. J. Oncol. 1998, 13, 893–906. [Google Scholar] [CrossRef]
- Andreasen, P.A.; Egelund, R.; Petersen, H.H. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol. Life Sci. 2000, 57, 25–40. [Google Scholar] [CrossRef]
- Harbeck, N.; Schmitt, M.; Meisner, C.; Friedel, C.; Untch, M.; Sweep, C.; Lisboa, B.; Lux, M.; Beck, T.; Hasmüller, S.; et al. Ten-year analysis of the prospective multicentre Chemo-N0 trial validates American Society of Clinical Oncology (ASCO)-recommended biomarkers uPA and PAI-1 for therapy decision making in node-negative breast cancer patients. Eur. J. Cancer 2013, 49, 1825–1835. [Google Scholar] [CrossRef]
- Nekarda, H.; Siewert, J.R.; Schmitt, M.; Ulm, K. Tumour-associated proteolytic factors uPA and PAI-1 and survival in totally resected gastric cancer. Lancet 1994, 343, 117. [Google Scholar] [CrossRef]
- Noh, H.; Hong, S.; Huang, S. Role of urokinase receptor in tumor progression and development. Theranostics 2013, 3, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Wosikowski, K. Inhibitory Properties of WX-UK1 on the Viability of Various Tumor Cell Lines; 50.C7.01.01; Wilex AG: Munich, Germany, 2001. [Google Scholar]
- Foekens, J. Anti-Tumor Efficacy Study with WX-UK1 in Rat Pancreatic Tumor Model CA90248 (Pancwilex 03); Erasmus University Medical Center Rotterdam: Rotterdam, The Netherlands, 2001. [Google Scholar]
- Foekens, J. Anti-Tumor Efficacy Study with L-WX-UK1 in Rat Mammary Tumor (Mamwilex 05); Erasmus University Medical Center Rotterdam: Rotterdam, The Netherlands, 2001. [Google Scholar]
- Foekens, J. Anti-Tumor Efficacy Study with WX-UK1 in Rat Mammary Tumor BN 472: Effect of Dosing Frequency no. 1 (Mamwilex 15); Erasmus University Medical Center Rotterdam: Rotterdam, The Netherlands, 2002. [Google Scholar]
- Krueger, A. Effects of WX-UK1 on the Formulation of Liver Metastases in Female DBA/2 Mice in the Syngeneic T-Cell Lymphoma L-Cl.5s Mouse Model; Technical University Munich: Munich, Germany, 2001. [Google Scholar]
- Paprottka, P.M.; Roßpunt, S.; Ingrisch, M.; Cyran, C.C.; Nikolaou, K.; Reiser, M.F.; Mack, B.; Gires, O.; A Clevert, D.; Zengel, P. Reducing tumor growth and angiogenesis using a triple therapy measured with Contrast-enhanced ultrasound (CEUS). BMC Cancer 2015, 15, 373. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, N.; Grodzki, A.C.; Gorin, F. Mis-trafficking of endosomal urokinase proteins triggers drug-induced glioma nonapoptotic cell death. Mol. Pharmacol. 2015, 87, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Guozhong, J.; Cao, F.; Ren, G.; Gao, D.; Bhakta, V.; Zhang, Y.; Cao, H.; Dong, Z.; Zang, W.; Zhang, S.; et al. PRSS3 Promotes Tumour Growth and Metastasis of Human Pancreatic Cancer. Gut 2010, 59, 1535–1544. [Google Scholar]
- Oldenburg, E.; Schar, C.R.; Lange, E.L.; Plasse, T.F.; Abramson, D.T.; Levitt, M.L.; Fathi, R.; Jensen, J.K. Abstract B055: New potential therapeutic applications of WX-UK1, as a specific and potent inhibitor of human trypsin-2 and human trypsin-3. Mol. Cancer Ther. 2018, 17, B055. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F2 Gen | Implanted Tumor | |||
|---|---|---|---|---|
| Marker | Allele No. 1 | Allele No. 2 | Allele No. 1 | Allele No. 2 |
| AMEL | X | X | X | X |
| D3S1358 | 14 | 15 | 14 | 15 |
| D1S1656 | 15 | 16 | 15 | 16 |
| D2S441 | 14 | 16 | 14 | 16 |
| D10S1248 | 16 | 16 | 16 | 16 |
| D13S317 | 8 | 8 | 8 | 8 |
| Penta E | 12 | 14 | 12 | 14 |
| D16S539 | 12 | 13 | 13 | 13 |
| D18S51 | 13 | 13 | 13 | 13 |
| D2S1338 | 17 | 23 | 17 | 23 |
| CSF1PO | 11 | 12 | 11 | 12 |
| Penta D | 9 | 10 | 9 | 10 |
| TH01 | 7 | 7 | 7 | 7 |
| vWA | 18 | 20 | 18 | 20 |
| D21S11 | 29 | 31.2 | 29 | 31.2 |
| D7S820 | 8 | 8 | 8 | 8 |
| D5S818 | 12 | 12 | 12 | 12 |
| TPOX | 8 | 8 | 8 | 8 |
| D8S1179 | 13 | 15 | 13 | 15 |
| D12S391 | 17 | 18 | 17 | 18 |
| D19S433 | 14 | 15 | 14 | 15 |
| FGA | 21 | 23 | 21 | 23 |
| D22S1045 | 16 | 17 | 16 | 17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asumda, F.Z.; Campbell, N.A.; Hassan, M.A.; Fathi, R.; Vasquez Rico, D.F.; Kiem, M.; Vang, E.V.; Kim, Y.H.; Luo, X.; O’Brien, D.R.; et al. Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts. Cancers 2024, 16, 1050. https://doi.org/10.3390/cancers16051050
Asumda FZ, Campbell NA, Hassan MA, Fathi R, Vasquez Rico DF, Kiem M, Vang EV, Kim YH, Luo X, O’Brien DR, et al. Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts. Cancers. 2024; 16(5):1050. https://doi.org/10.3390/cancers16051050
Chicago/Turabian StyleAsumda, Faizal Z., Nellie A. Campbell, Mohamed A. Hassan, Reza Fathi, Daniella F. Vasquez Rico, Melanie Kiem, Ethan V. Vang, Yo Han Kim, Xin Luo, Daniel R. O’Brien, and et al. 2024. "Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts" Cancers 16, no. 5: 1050. https://doi.org/10.3390/cancers16051050
APA StyleAsumda, F. Z., Campbell, N. A., Hassan, M. A., Fathi, R., Vasquez Rico, D. F., Kiem, M., Vang, E. V., Kim, Y. H., Luo, X., O’Brien, D. R., Buhrow, S. A., Reid, J. M., Moore, M. J., Ben-Yair, V. K., Levitt, M. L., Leiting, J. L., Abdelrahman, A. M., Zhu, X., Lucien, F., ... Roberts, L. R. (2024). Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts. Cancers, 16(5), 1050. https://doi.org/10.3390/cancers16051050