
Recent Advances in Pathology of Intrahepatic Cholangiocarcinoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Risk Factors
3. Cells of Origin
4. Clinical Features
5. Radiological Features
6. Molecular Features
7. Immunohistochemical Features
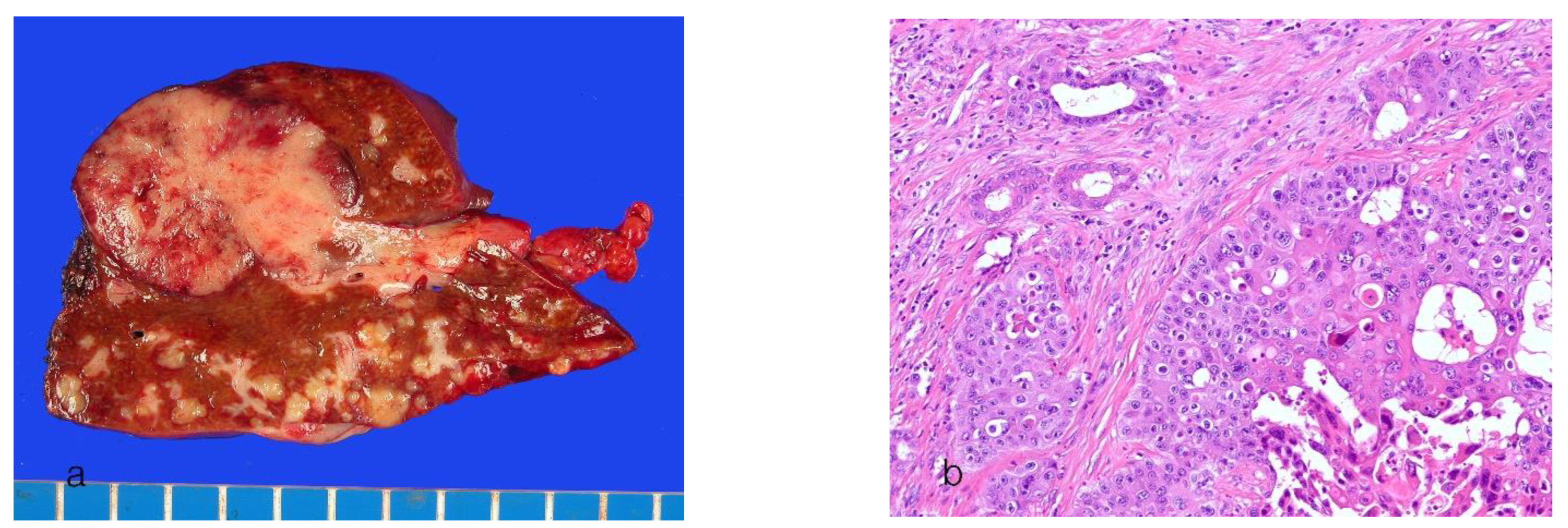
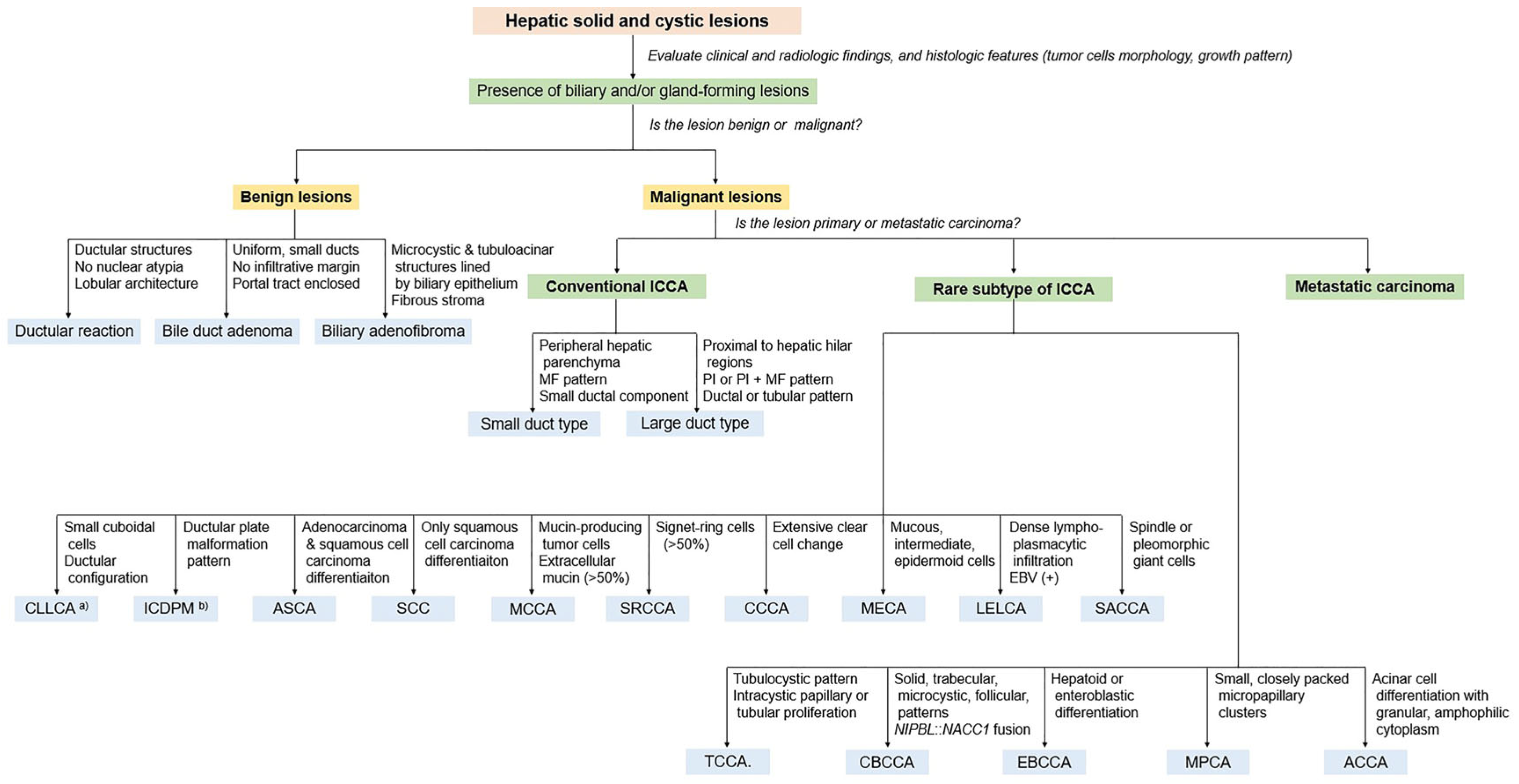
8. Pathological Features of Conventional Intrahepatic Cholangiocarcinoma
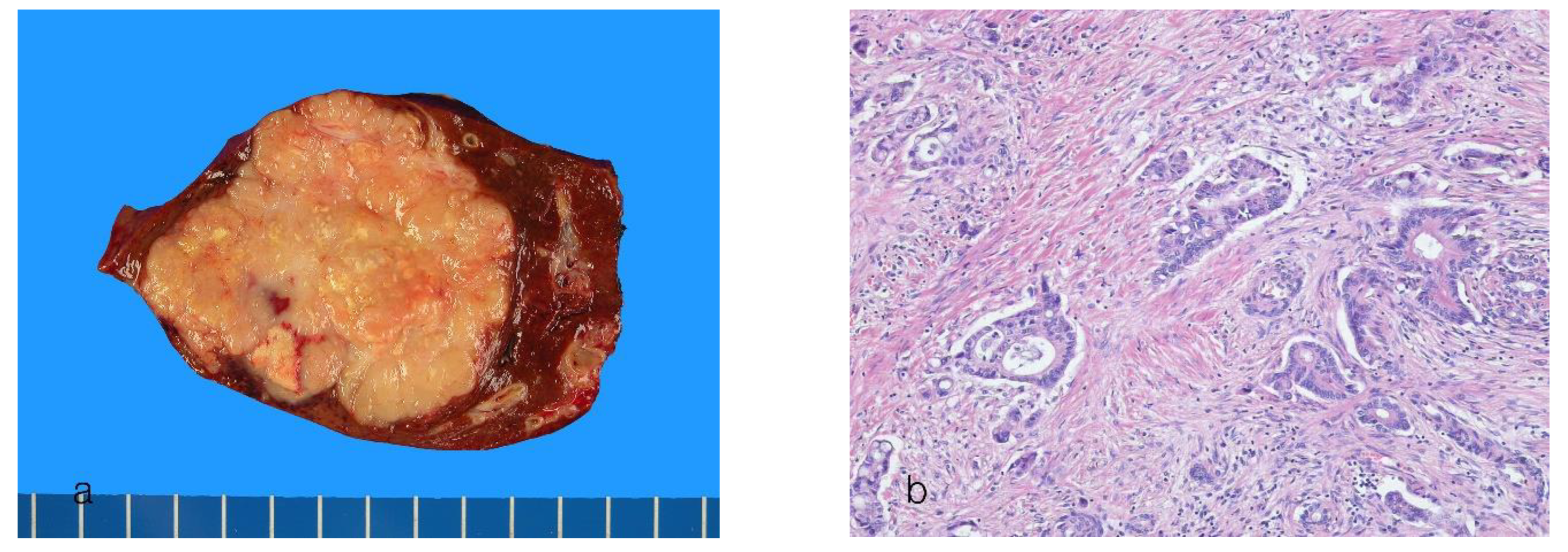
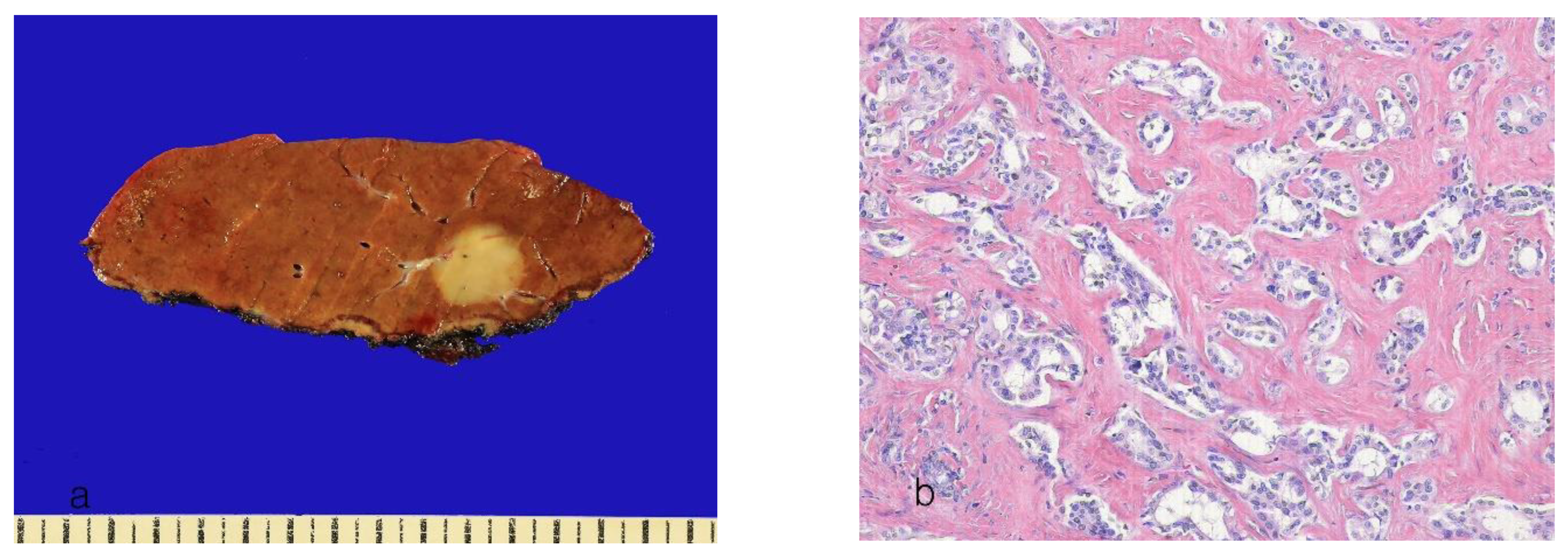
8.1. Small-Duct Intrahepatic Cholangiocarcinoma
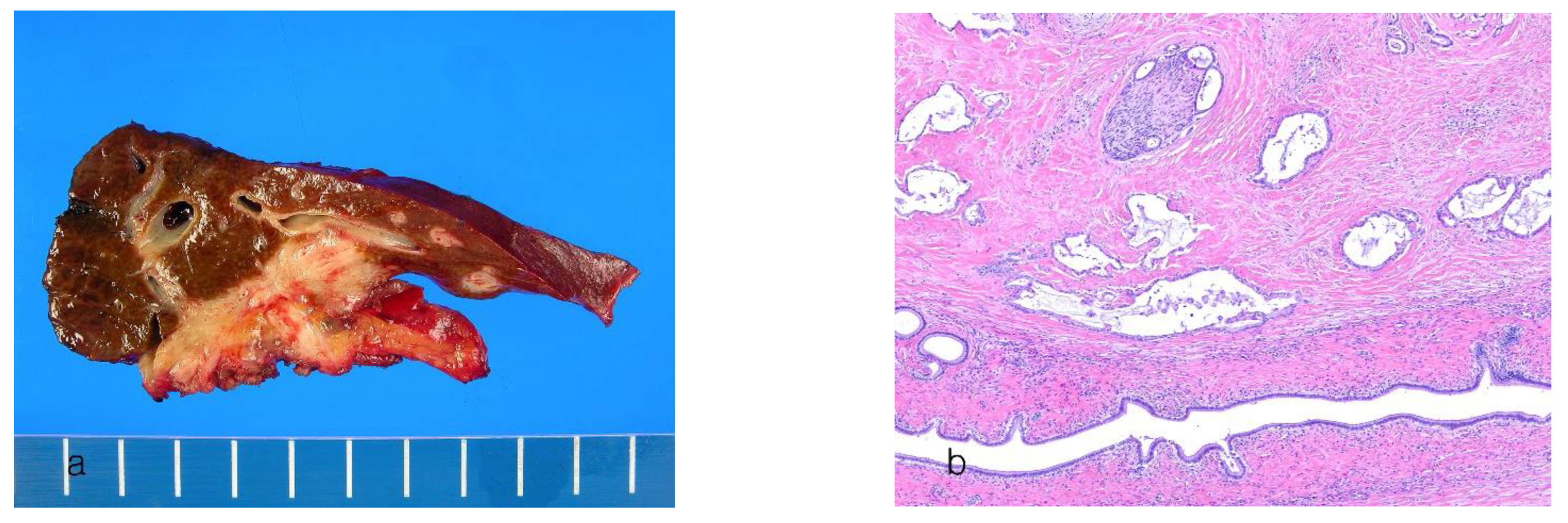
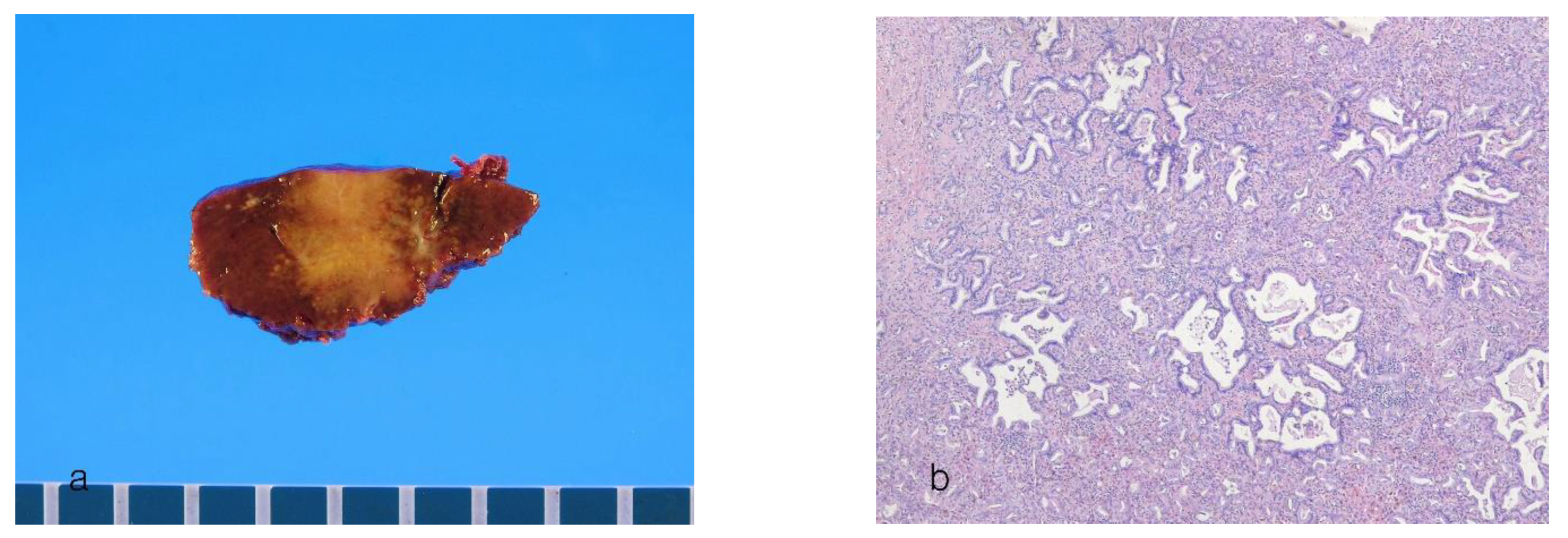
8.2. Large-Duct Intrahepatic Cholangiocarcinoma
8.3. Histological Grading
8.4. Premalignant Lesions of Intrahepatic Cholangiocarcinoma
9. Rare Subtypes of Intrahepatic Cholangiocarcinoma
9.1. Cholangiolocarcinoma
9.2. ICCA with Ductal Plate Malformation
9.3. Adenosquamous Carcinoma
9.4. Squamous Cell Carcinoma
9.5. Mucinous Carcinoma
9.6. Signet Ring Cell Carcinoma
9.7. Clear Cell Cholangiocarcinoma
9.8. Mucoepidermoid Carcinoma
9.9. Lymphoepithelioma-like Cholangiocarcinoma
9.10. Sarcomatous Cholangiocarcinoma
10. New Provisional Subtypes of Intrahepatic Cholangiocarcinoma Not Included in the 2019 WHO Classification
10.1. Tubulocystic Carcinoma of the Bile Duct
10.2. Cholangioblastic Cholangiocarcinoma
10.3. Enteroblastic Cholangiocarcinomae
10.4. Micropapillary Carcinoma
10.5. Acinar Cell Carcinoma
11. Pathological Diagnostic Approach
11.1. Specimen Handling
11.1.1. Biopsy Specimens
11.1.2. Hepatectomy Specimens
11.2. Pathological Diagnostic Approach
12. Future Perspectives
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nakanuma, Y.; Klimstra, D.S.; Komuta, M.; Zen, Y. Intrahepatic cholangiocarcinoma. In WHO Classification of Tumours. Digestive System Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2019; pp. 254–258. ISBN 978-92-832-4499-8.2. [Google Scholar]
- Tyson, G.L.; Ilyas, J.A.; Duan, Z.; Green, L.K.; Younes, M.; El-Serag, H.B.; Davila, J.A. Secular trends in the incidence of cholangiocarcinoma in the USA and the impact of misclassification. Dig. Dis. Sci. 2014, 59, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-year trends in cholangiocarcinoma incidence in the U.S.: Intrahepatic disease on the rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Antwi, S.O.; Mousa, O.Y.; Patel, T. Racial, ethnic, and age disparities in incidence and survival of intrahepatic cholangiocarcinoma in the United States; 1995–2014. Ann. Hepatol. 2018, 17, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 7–18. [Google Scholar] [CrossRef] [PubMed]
- Aishima, S.; Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: Different characters of perihilar large duct type versus peripheral small duct type. J. Hepatobiliary Pancreat. Sci. 2015, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.S.; Raza, A.; Propst, R.; Adeyi, O.; Bateman, J.; Sopha, S.C.; Shaw, J.; Auerbach, A. Recent advances in digestive tract tumors: Updates from the 5th edition of the World Health Organization “Blue Book”. Arch. Pathol. Lab. Med. 2021, 145, 607–626. [Google Scholar] [CrossRef]
- Gopal, P.; Robert, M.E.; Zhang, X. Cholangiocarcinoma: Pathologic and molecular classification in the era of precision medicine. Arch. Pathol. Lab. Med. 2023, 148, 359–370. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Féliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.; Park, Y.N. Up-to-date pathologic classification and molecular characteristics of intrahepatic cholangiocarcinoma. Front. Med. 2022, 9, 857140. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.; Papoutsoglou, P.; Coulouarn, C. Molecular classification of cholangiocarcinoma. Curr. Opin. Gastroenterol. 2020, 36, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dixon, E. Epidemiology and risk factors: Intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Mori, T.; Yokoyama, M.; Nakazato, T.; Abe, N.; Nakanuma, Y.; Tsubouchi, H.; Sugiyama, M. Hepatolithiasis: Analysis of Japanese nationwide surveys over a period of 40 years. J. Hepatobiliary Pancreat. Sci. 2014, 21, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.K.; Chaudhuri, B.; Schuler, J.J.; Nyhus, L.M. Carcinoma associated with congenital cystic dilation of bile ducts. Arch. Surg. 1982, 117, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Sameshima, Y.; Uchimura, M.; Muto, Y.; Maeda, J.; Tsuchiyama, H. Coexistent carcinoma in congenital dilatation of the bile duct and anomalous arrangement of the pancreatico-bile duct. Carcinogenesis of coexistent gall bladder carcinoma. Cancer 1987, 60, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Rubel, L.R.; Ishak, K.G. Thorotrast-associated cholangiocarcinoma: An epidemiologic and clinicopathologic study. Cancer 1982, 50, 1408–1415. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Olaizola, P.; Paiva, N.A.; Olaizola, I.; Agirre-Lizaso, A.; Landa, A.; Bujanda, L.; Perugorria, M.J.; Banales, J.M. Pathogenesis of Cholangiocarcinoma. Annu. Rev. Pathol. 2021, 16, 433–463. [Google Scholar] [CrossRef]
- Komuta, M.; Govaere, O.; Vandecaveye, V.; Akiba, J.; Van Steenbergen, W.; Verslype, C.; Laleman, W.; Pirenne, J.; Aerts, R.; Yano, H.; et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 2012, 55, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Investig. 2012, 122, 3914–3918. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Komuta, M.; Spee, B.; Vander Borght, S.; De Vos, R.; Verslype, C.; Aerts, R.; Yano, H.; Suzuki, T.; Matsuda, M.; Fujii, H.; et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 2008, 47, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, P.; Preiss, J.; Sasatomi, E.; Gerber, D.A. Biliary adenofibroma: A rare hepatic lesion with malignant features. Hepatology 2017, 65, 380–383. [Google Scholar] [CrossRef]
- Bhalla, A.; Mann, S.A.; Chen, S.; Cummings, O.W.; Lin, J. Histopathological evidence of neoplastic progression of von Meyenburg complex to intrahepatic cholangiocarcinoma. Hum. Pathol. 2017, 67, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Wang, Y.; Carpino, G.; Reid, L.M.; Gaudio, E.; Alvaro, D. Mucin-producing cholangiocarcinoma might derive from biliary tree stem/progenitor cells located in peribiliary glands. Hepatology 2012, 55, 2041–2042. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Sudo, Y. Biliary tumors with pancreatic counterparts. Semin. Diagn. Pathol. 2017, 34, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Uchida, T.; Sato, Y.; Uesaka, K. An S100P-positive biliary epithelial field is a preinvasive intraepithelial neoplasm in nodular-sclerosing cholangiocarcinoma. Hum. Pathol. 2017, 60, 46–57. [Google Scholar] [CrossRef]
- Nevi, L.; Di Matteo, S.; Carpino, G.; Zizzari, I.G.; Samira, S.; Ambrosino, V.; Costantini, D.; Overi, D.; Giancotti, A.; Monti, M.; et al. DCLK1, a putative stem cell marker in human cholangiocarcinoma. Hepatology 2021, 73, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Joo, I.; Lee, J.M.; Yoon, J.H. Imaging diagnosis of intrahepatic and perihilar cholangiocarcinoma: Recent advances and challenges. Radiology 2018, 288, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Rimola, J.; Forner, A.; Reig, M.; Vilana, R.; de Lope, C.R.; Ayuso, C.; Bruix, J. Cholangiocarcinoma in cirrhosis: Absence of contrast washout in delayed phases by magnetic resonance imaging avoids misdiagnosis of hepatocellular carcinoma. Hepatology 2009, 50, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Galassi, M.; Iavarone, M.; Rossi, S.; Bota, S.; Vavassori, S.; Rosa, L.; Leoni, S.; Venerandi, L.; Marinelli, S.; Sangiovanni, A.; et al. Patterns of appearance and risk of misdiagnosis of intrahepatic cholangiocarcinoma in cirrhosis at contrast enhanced ultrasound. Liver Int. 2013, 33, 771–799. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, V.; Fowler, K.J.; Kamaya, A.; Kielar, A.Z.; Elsayes, K.M.; Bashir, M.R.; Kono, Y.; Do, R.K.; Mitchell, D.G.; Singal, A.G.; et al. Liver imaging reporting and data system (LI-RADS) version 2018: Imaging of hepatocellular carcinoma in at-risk patients. Radiology 2018, 289, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Barriuso, J.; Chander, A.; McNamara, M.G.; Hubner, R.A.; ÓReilly, D.; Manoharan, P.; Valle, J.W. 18F-fluorodeoxyglucose positron emission tomography (18FDG-PET) for patients with biliary tract cancer: Systematic review and meta-analysis. J. Hepatol. 2019, 71, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Fujikura, K.; Ajiki, T.; Fukumoto, T.; Otani, K.; Azuma, T.; Itoh, T.; Ku, Y.; Zen, Y. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod. Pathol. 2017, 30, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zen, Y. Intrahepatic cholangiocarcinoma: Typical features, uncommon variants, and controversial related entities. Hum. Pathol. 2023, 132, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Misumi, K.; Shibahara, J.; Arita, J.; Sakamoto, Y.; Hasegawa, K.; Kokudo, N.; Fukayama, M. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 2016, 40, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Tsai, J.H.; Yuan, R.H.; Chang, C.N.; Lee, H.J.; Jeng, Y.M. Morphological subclassification of intrahepatic cholangiocarcinoma: Etiological, clinicopathological, and molecular features. Mod. Pathol. 2014, 27, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Akita, M.; Sung, Y.N.; Fujikura, K.; Lee, J.H.; Hwang, S.; Yu, E.; Otani, K.; Hong, S.M.; Zen, Y. MDM2 amplification in intrahepatic cholangiocarcinomas: Its relationship with large-duct type morphology and uncommon KRAS mutations. Am. J. Surg. Pathol. 2018, 42, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Sasaki, T.; Serikawa, M.; Minami, T.; Okazaki, A.; Yukutake, M.; Ishigaki, T.; Kosaka, K.; Mouri, T.; Yoshimi, S.; et al. Elevated expression of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 in primary sclerosing cholangitis: Implications for cholangiocarcinogenesis. Int. J. Oncol. 2013, 43, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Sasaki, M.; Igarashi, S.; Sato, Y.; Nakanuma, Y. KRAS and GNAS mutations and p53 overexpression in biliary intraepithelial neoplasia and intrahepatic cholangiocarcinomas. Cancer 2013, 119, 1669–1674. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, H.; Zhou, D.; Wang, H.; Wang, Q.; Zou, S.; Tu, Q.; Wu, M.; Hu, H. Hepatitis B virus-associated intrahepatic cholangiocarcinoma and hepatocellular carcinoma may hold common disease process for carcinogenesis. Eur. J. Cancer 2010, 46, 1056–1061. [Google Scholar] [CrossRef]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Suda, R.; Sakai, N.; Matsushita, K.; Ishige, T.; Kawasaki, Y.; Shiko, Y.; Furukawa, K.; Mishima, T.; Nakadai, E.; Ohtsuka, M. Prediction of mismatch repair deficient biliary tract cancer: Role of morphological features and host immune response detected by routine hematoxylin-eosin staining. J. Hepatobiliary Pancreat. Sci. 2021, 28, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.Y.; Dibbern, M.E.; Mahadevan, M.S.; Fan, J.; Kunk, P.R.; Stelow, E.B. Mismatch repair protein deficiency/microsatellite instability is rare in cholangiocarcinomas and associated with distinctive morphologies. Am. J. Clin. Pathol. 2020, 153, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Lee, J.H.; Yoon, J.H.; Kim, C.Y.; Lee, H.S.; Jang, J.J.; Yi, N.J.; Suh, K.S.; Lee, K.U.; Park, E.S.; et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010, 70, 3034–3041. [Google Scholar] [CrossRef] [PubMed]
- Montpetit, A.; Boily, G.; Sinnett, D. A detailed transcriptional map of the chromosome 12p12 tumour suppressor locus. Eur. J. Hum. Genet. 2002, 10, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Akita, M.; Sawada, R.; Komatsu, M.; Suleman, N.; Itoh, T.; Ajiki, T.; Heaton, N.; Fukumoto, T.; Zen, Y. An immunostaining panel of C-reactive protein, N-cadherin, and S100 calcium binding protein P is useful for intrahepatic cholangiocarcinoma subtyping. Hum. Pathol. 2021, 109, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Lei, H.J.; Chen, M.H.; Ho, H.L.; Chiu, L.Y.; Li, C.P.; Wang, Y.C. C-reactive protein (CRP) is a promising diagnostic immunohistochemical marker for intrahepatic cholangiocarcinoma and is associated with better prognosis. Am. J. Surg. Pathol. 2017, 41, 1630–1641. [Google Scholar] [CrossRef]
- Zen, Y.; Britton, D.; Mitra, V.; Pike, I.; Sarker, D.; Itoh, T.; Heaton, N.; Quaglia, A. Tubulin beta-III: A novel immunohistochemical marker for intrahepatic peripheral cholangiocarcinoma. Histopathology 2014, 65, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Sofue, K.; Fujikura, K.; Otani, K.; Itoh, T.; Ajiki, T.; Fukumoto, T.; Zen, Y. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB 2019, 21, 226–234. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Ting, D.T.; Shahid, M.; Konstantinidis, I.T.; Sabbatino, F.; Goyal, L.; Rice-Stitt, T.; Mubeen, A.; Arora, K.; Bardeesey, N.; et al. The ability to diagnose intrahepatic cholangiocarcinoma definitively using novel branched DNA-enhanced albumin RNA in situ hybridization technology. Ann. Surg. Oncol. 2016, 23, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Sigel, C.S.; Drill, E.; Zhou, Y.; Basturk, O.; Askan, G.; Pak, L.M.; Vakiani, E.; Wang, T.; Boerner, T.; Do, R.K.G.; et al. Intrahepatic cholangiocarcinomas have histologically and immunophenotypically distinct small and large duct patterns. Am. J. Surg. Pathol. 2018, 42, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Kokudo, N.; Matsuyama, Y.; Sakamoto, M.; Izumi, N.; Kadoya, M.; Kaneko, S.; Ku, Y.; Kudo, M.; Takayama, T.; et al. Proposal of a new staging system for intrahepatic cholangiocarcinoma: Analysis of surgical patients from a nationwide survey of the Liver Cancer Study Group of Japan. Cancer 2016, 122, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Kakuda, Y. Pathologic classification of cholangiocarcinoma: New concepts. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M.; Zen, Y.; Yeh, M.M. Tumors of the Livers, AFIP Atlas of Tumor Pathology; 4th series; American Registry of Pathology: Washington, DC, USA, 2018; Volume 27, pp. 201–254. ISBN 978-1-933477-41-1. [Google Scholar]
- Nakanuma, Y.; Miyata, T.; Uchida, T. Latest advances in the pathological understanding of cholangiocarcinomas. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 113–127. [Google Scholar] [CrossRef]
- Uno, M.; Shimada, K.; Yamamoto, Y.; Nara, S.; Esaki, M.; Sakamoto, Y.; Kosuge, T.; Ojima, H. Periductal infiltrating type of intrahepatic cholangiocarcinoma: A rare macroscopic type without any apparent mass. Surg. Today 2012, 42, 1189–1194. [Google Scholar] [CrossRef]
- Lawrence, J.; Burgart, L.J.; Chopp, W.V.; Dhanpat, J. Protocol for the Examination of Specimens from Patients with Carcinoma of the Intrahepatic Bile Ducts. College of American Pathologists. Available online: https://documents.cap.org/protocols/BileDuctIH_4.2.0.0.REL_CAPCP.pdf?_gl=1*rg0bxi*ga*NDU0NDI1NTg2LjE2ODk2NzA2MDM.*_ga_97ZFJSQQ0X*MTcwMzU1NTQxMy4yLjAuMTcwMzU1NTQxOC4wLjAuMA (accessed on 25 December 2023).
- Zen, Y.; Adsay, N.V.; Bardadin, K.; Colombari, R.; Ferrell, L.; Haga, H.; Hong, S.M.; Hytiroglou, P.; Klöppel, G.; Lauwers, G.Y.; et al. Biliary intraepithelial neoplasia: An international interobserver agreement study and proposal for diagnostic criteria. Mod. Pathol. 2007, 20, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Basturk, O.; Esposito, I.; Klimstra, D.S.; Komuta, M.; Zen, Y. Intraductal papillary neoplasm of the bile ducts. In WHO Classification of Tumours. Digestive System Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2019; pp. 279–282. ISBN 978-92-832-4499-82.2.2. [Google Scholar]
- Hoang, M.P.; Murakata, L.A.; Katabi, N.; Henson, D.E.; Albores-Saavedra, J. Invasive papillary carcinomas of the extrahepatic bile ducts: A clinicopathologic and immunohistochemical study of 13 cases. Mod. Pathol. 2002, 15, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Jang, K.T.; Fukushima, N.; Furukawa, T.; Hong, S.M.; Kim, H.; Lee, K.B.; Zen, Y.; Jang, J.Y.; Kubota, K. A statement by the Japan-Korea expert pathologists for future clinicopathological and molecular analyses toward consensus building of intraductal papillary neoplasm of the bile duct through several opinions at the present stage. J. Hepatobiliary Pancreat. Sci. 2018, 25, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Pehlivanoglu, B.; Adsay, V. Intraductal tubulopapillary neoplasms of the bile ducts: Identity, clinicopathologic characteristics, and differential diagnosis of a distinct entity among intraductal tumors. Hum. Pathol. 2023, 132, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, A.M.; Jang, K.T.; Klöppel, G.; Saka, B.; Hong, S.M.; Choi, H.; Offerhaus, G.J.; Hruban, R.H.; Zen, Y.; Konukiewitz, B.; et al. Intraductal tubulopapillary neoplasms of the bile ducts: Clinicopathologic, immunohistochemical, and molecular analysis of 20 cases. Mod. Pathol. 2015, 28, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, T.; Sakamoto, M.; Mukai, K.; Kawano, N.; Konishi, M.; Ryu, M.; Fukamachi, S.; Hirohashi, S. Cholangiocarcinoma arising in bile duct adenoma with focal area of bile duct hamartoma. Virchows Arch. 1995, 426, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Pujals, A.; Amaddeo, G.; Castain, C.; Bioulac-Sage, P.; Compagnon, P.; Zucman-Rossi, J.; Azoulay, D.; Leroy, K.; Zafrani, E.S.; Calderaro, J. BRAF V600E mutations in bile duct adenomas. Hepatology 2015, 61, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Steiner, P.E.; Higginson, J. Cholangiolocellular carcinoma of the liver. Cancer 1959, 12, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Theise, N.D.; Nakashima, O.; Park, Y.N.; Nakanuma, Y. Combined hepatocellular-cholangiocarcinoma. In WHO Classification of Tumours of Digestive System, 4th ed.; Bosman, F.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; IARC Press: Lyon, France, 2010; pp. 225–227. ISBN 978-92-832-4499-8. [Google Scholar]
- Ariizumi, S.; Kotera, Y.; Katagiri, S.; Nakano, M.; Nakanuma, Y.; Saito, A.; Yamamoto, M. Long-term survival of patients with cholangiolocellular carcinoma after curative hepatectomy. Ann. Surg. Oncol. 2014, 21 (Suppl. S3), S451–S458. [Google Scholar] [CrossRef]
- Kozaka, K.; Sasaki, M.; Fujii, T.; Harada, K.; Zen, Y.; Sato, Y.; Sawada, S.; Minato, H.; Matsui, O.; Nakanuma, Y. A subgroup of intrahepatic cholangiocarcinoma with an infiltrating replacement growth pattern and a resemblance to reactive proliferating bile ductules: ‘bile ductular carcinoma’. Histopathology 2007, 51, 390–400. [Google Scholar] [CrossRef]
- Westerhoff, M.; Lamps, L.W.; Kakar, S. Intrahepatic cholangiocarcinoma. In Diagnostic Pathology. Hepatobiliary and Pancreas, 3rd ed.; Elsevier: Philadelphia, PA, USA, 2022; pp. 310–311. ISBN 978-0-323-77620-2. [Google Scholar]
- Nakanuma, Y.; Sato, Y.; Ikeda, H.; Harada, K.; Kobayashi, M.; Sano, K.; Uehara, T.; Yamamoto, M.; Ariizumi, S.; Park, Y.N.; et al. Intrahepatic cholangiocarcinoma with predominant “ductal plate malformation” pattern: A new subtype. Am. J. Surg. Pathol. 2012, 36, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.; Rhee, H.; Shim, H.S.; Yoo, J.E.; Choi, G.H.; Kim, H.; Park, Y.N. Genetic, clinicopathological, and radiological features of intrahepatic cholangiocarcinoma with ductal plate malformation pattern. Gut Liver 2022, 16, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, M.; Lamps, L.W.; Kakar, S. Von Meyenburg complex (biliary microhamartoma). In Diagnostic Pathology. Hepatobiliary and Pancreas, 3rd ed.; Elsevier: Philadelphia, PA, USA, 2022; pp. 304–305. ISBN 978-0-323-77620-2. [Google Scholar]
- Tsui, W.M.; Nakanuma, Y. Biliary adenofibroma. In WHO Classification of Tumours. Digestive System Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2019; pp. 248–249. ISBN 978-92-832-4499-8. [Google Scholar]
- Kobayashi, M.; Okabayashi, T.; Okamoto, K.; Namikawa, T.; Araki, K. A clinicopathologic study of primary adenosquamous carcinoma of the liver. J. Clin. Gastroenterol. 2005, 39, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.N.; Jan, Y.Y.; Chen, M.F. Adenosquamous carcinoma of the liver: Clinicopathologic features in 12 patients and review of the literature. Int. Surg. 2002, 87, 125–129. [Google Scholar] [PubMed]
- Maeda, T.; Takenaka, K.; Taguchi, K.; Kajiyama, K.; Shirabe, K.; Shimada, M.; Tsuneyoshi, M.; Sugimachi, K. Adenosquamous carcinoma of the liver: Clinicopathologic characteristics and cytokeratin profile. Cancer 1997, 80, 364–371. [Google Scholar] [CrossRef]
- Nakajima, T.; Kondo, Y.; Miyazaki, M.; Okui, K. A histopathologic study of 102 cases of intrahepatic cholangiocarcinoma: Histologic classification and modes of spreading. Hum. Pathol. 1988, 19, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, G. Primary squamous cell carcinoma of the liver: A case report. J. Int. Med. Res. 2021, 49, 3000605211021275. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, H.; Azhdari Tehrani, H.; Salari, S.; Feiziazar, S.; Darnahal, M. Primary squamous cell carcinoma of the liver: A case report. Gastroenterol. Hepatol. Bed. Bench 2022, 15, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Bloustein, P.A.; Silverberg, S.G. Squamous cell carcinoma originating in an hepatic cyst. Case report with a review of the hepatic cyst-carcinoma association. Cancer 1976, 38, 2002–2005. [Google Scholar] [CrossRef]
- Lynch, M.J.; McLeod, M.K.; Weatherbee, L.; Gilsdorf, J.R.; Guice, K.S.; Eckhauser, F.E. Squamous cell cancer of the liver arising from a solitary benign nonparasitic hepatic cyst. Am. J. Gastroenterol. 1988, 83, 426–431. [Google Scholar]
- Zhang, X.F.; Du, Z.Q.; Liu, X.M.; Lv, Y. Primary squamous cell carcinoma of liver: Case series and review of literatures. Medicine 2015, 94, e868. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Jan, Y.Y.; Chen, T.C. Clinical studies of mucin-producing cholangiocellular carcinoma: A study of 22 histopathology-proven cases. Ann. Surg. 1998, 227, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Ahuja, A.T.; Kwong, K.H.; Fung, K.S.; Lai, C.K.; Lau, J.W. Mucinous cholangiocarcinoma: An unusual complication of hepatolithiasis and recurrent pyogenic cholangitis. Histopathology 1997, 30, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Matsui, O.; Ueda, K.; Kadoya, M.; Yoshikawa, J.; Gabata, T.; Takashima, T.; Izumi, R.; Nakanuma, Y. Imaging findings of mucinous type of cholangiocellular carcinoma. J. Comput. Assist. Tomogr. 1996, 20, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Ohta, H.; Arisato, S.; Nakano, Y.; Murakami, M.; Orii, Y.; Saito, H.; Sakurai, Y.; Sakurai, H.; Sato, T.; et al. Case report: Mucinous cholangiocarcinoma featuring a multicystic appearance and periportal collar in imaging. J. Gastroenterol. Hepatol. 1999, 4, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Chan, C.W. Mucin-producing cholangiocarcinoma: An autopsy study in Hong Kong. Pathology 1976, 8, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Masaki, Y.; Akutsu, N.; Adachi, Y.; Ishigami, K.; Iwata, N.; Endo, T.; Ishii, Y.; Sasaki, Y.; Nagayama, M.; Kimura, Y.; et al. Genomic analysis of an aggressive case with metastatic intrahepatic mucinous cholangiocarcinoma. Clin. J. Gastroenterol 2022, 15, 809–817. [Google Scholar] [CrossRef]
- Zeng, X.; Ou, H.; Zeng, C.; Liu, Q.; Wang, W.; Yao, J. Multi-omics integrated analyzed the origin of intrahepatic mucinous cholangiocarcinoma: A case report. Front. Oncol. 2023, 13, 1175707. [Google Scholar] [CrossRef] [PubMed]
- Zen, Y.; Quaglia, A.; Heaton, N.; Rela, M.; Portmann, B. Two distinct pathways of carcinogenesis in primary sclerosing cholangitis. Histopathology 2011, 59, 1100–1110. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Curado, M.P.; Franceschi, S.; Gores, G.; Paradis, V.; Sripa, B.; Tsui, W.M.S.; Wee, A. Intrahepatic cholangiocarcinoma. In WHO Classification of Tumours of Digestive System, 4th ed.; Bosman, F.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; IARC Press: Lyon, France, 2010; pp. 217–224. ISBN 978-92-832-4499-8. [Google Scholar]
- Zen, Y.; Fujii, T.; Itatsu, K.; Nakamura, K.; Minato, H.; Kasashima, S.; Kurumaya, H.; Katayanagi, K.; Kawashima, A.; Masuda, S.; et al. Biliary papillary tumors share pathological features with intraductal papillary mucinous neoplasm of the pancreas. Hepatology 2006, 44, 1333–1343. [Google Scholar] [CrossRef]
- Saito, K.; Nakanuma, Y. Signet ring cell carcinoma of the intrahepatic bile duct. Ryoikibetsu Shokogun Shirizu 1995, 7, 395–397. [Google Scholar]
- Younes, R.; Ponzo, P.; Marzano, A. Microangiopathic hemolytic anemia caused by a signet-ring cell carcinoma of the intrahepatic bile duct. Minerva. Gastroenterol. Dietol. 2017, 63, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Kim, C.; Kim, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; Chung, J.B.; Kim, H.; Bang, S. Signet ring cell carcinoma of the extrahepatic bile duct. Gut Liver 2010, 4, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Chedid, M.F.; Lucas, E.T.; Cerski, C.T.; Lopes, M.F.; Amaral, O.B.; Chedid, A.D. Signet-ring cell hilar cholangiocarcinoma: Case report. Arq. Bras. Cir. Dig. 2015, 28, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.B.; Wu, Y.; Li, F.; Zhao, K.F.; Shi, R.S.; Huang, Q.; Ao, J.; Ke, D. Primary signet-ring cell carcinoma of the extrahepatic bile duct: A case report. World J. Gastrointest. Oncol. 2022, 14, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Gütgemann, I.; Wolff, M.; Fischer, H.P. Intrahepatic clear cell cholangiocarcinoma: Immunohistochemical aspects in a very rare type of cholangiocarcinoma. Am. J. Surg. Pathol. 2007, 31, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.R.; Baldaia, C.; Pinto Marques, H.; Tortosa, F.; Ramalho, F. Intrahepatic clear cell cholangiocarcinoma—An uncommon histologic subtype: Case report and literature review. Rev. Esp. Enferm. Dig. 2017, 109, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Tihan, T.; Blumgart, L.; Klimstra, D.S. Clear cell papillary carcinoma of the liver: An unusual variant of peripheral cholangiocarcinoma. Hum. Pathol. 1998, 29, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Khera, R.; Uppin, S.G.; Uppin, M.S.; Sundaram, C. Clear cell papillary cholangiocarcinoma: A case report with review of literature. Indian J. Pathol. Microbiol. 2014, 57, 105–108. [Google Scholar] [CrossRef]
- Falta, E.M.; Rubin, A.D.; Harris, J.A. Peripheral clear cell cholangiocarcinoma: A rare histologic variant. Am. Surg. 1999, 65, 592–595. [Google Scholar] [CrossRef]
- Logani, S.; Adsay, V. Clear cell cholangiocarcinoma of the liver is a morphologically distinctive entity. Hum. Pathol. 1998, 29, 1548–1549. [Google Scholar] [CrossRef] [PubMed]
- Albores-Saavedra, J.; Hoang, M.P.; Murakata, L.A.; Sinkre, P.; Yaziji, H. Atypical bile duct adenoma, clear cell type: A previously undescribed tumor of the liver. Am. J. Surg. Pathol. 2001, 25, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y.; Shimada, M.; Ikegami, T.; Kubo, T.; Imura, S.; Morine, Y.; Kanemura, H.; Mori, H. Mucoepidermoid carcinoma of the liver: Report of a rare case and review of the literature. Hepatol. Res. 2008, 38, 736–742. [Google Scholar] [CrossRef]
- Seethala, R.R.; Dacic, S.; Cieply, K.; Kelly, L.M.; Nikiforova, M.N. A reappraisal of the MECT1/MAML2 translocation in salivary mucoepidermoid carcinomas. Am. J. Surg. Pathol. 2010, 34, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Jee, K.J.; Persson, M.; Heikinheimo, K.; Passador-Santos, F.; Aro, K.; Knuutila, S.; Odell, E.W.; Mäkitie, A.; Sundelin, K.; Stenman, G.; et al. Genomic profiles and CRTC1-MAML2 fusion distinguish different subtypes of mucoepidermoid carcinoma. Mod. Pathol. 2013, 26, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Kai, K.; Tanikawa, K.; Hiraki, M.; Mizukami, N.; Aishima, S.; Nakano, T.; Yamamoto, H. Primary mucoepidermoid carcinoma of the liver with CRTC1-MAML2 fusion: A case report. Diagn. Pathol. 2019, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Su, X.; Cao, W.; Xu, L.; Zhang, R.; Huang, Z.; Wang, J.; Li, L.; Wu, L.; Liao, W. Whole-exome sequencing reveals the etiology of the rare primary hepatic mucoepidermoid carcinoma. Diagn. Pathol. 2021, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Q.; Li, B.; Li, Y.; Tian, X.Y.; Li, Z. Unusual mucoepidermoid carcinoma of the liver misdiagnosed as squamous cell carcinoma by intraoperative histological examination. Diagn. Pathol. 2014, 9, 24. [Google Scholar] [CrossRef]
- Choi, D.; Kim, H.; Lee, K.S.; Lee, K.G.; Park, C.K. Mucoepidermoid carcinoma of the liver diagnosed as a liver abscess: Report of a case. Surg. Today 2004, 34, 968–972. [Google Scholar] [CrossRef]
- Hsu, H.C.; Chen, C.C.; Huang, G.T.; Lee, P.H. Clonal Epstein-Barr virus associated cholangiocarcinoma with lymphoepithelioma-like component. Hum. Pathol. 1996, 27, 848–850. [Google Scholar] [CrossRef]
- Chen, T.C.; Ng, K.F.; Kuo, T. Intrahepatic cholangiocarcinoma with lymphoepithelioma-like component. Mod. Pathol. 2001, 14, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Jeng, Y.M.; Chen, C.L.; Hsu, H.C. Lymphoepithelioma-like cholangiocarcinoma: An Epstein-Barr virus-associated tumor. Am. J. Surg. Pathol. 2001, 25, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Szekely, E. Lymphoepithelioma-like cholangiocarcinoma (LELC) not associated with Epstein-Barr virus. Am. J. Surg. Pathol. 2001, 25, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.W.; Tong, J.H.; Sung, M.Y.; Lai, P.B.; To, K.F. Epstein-Barr virus-associated lymphoepithelioma-like cholangiocarcinoma: A rare variant of intrahepatic cholangiocarcinoma with favourable outcome. Histopathology 2014, 65, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Labgaa, I.; Stueck, A.; Ward, S.C. Lymphoepithelioma-like carcinoma in liver. Am. J. Pathol. 2017, 187, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Alpert, L.; Hart, J.; Chapman, C.; Pillai, A.A. Lymphoepithelioma-Like Carcinomas: A Rare Variant of Cholangiocarcinoma. Hepatology 2020, 72, 353–355. [Google Scholar] [CrossRef]
- Tsai, J.H.; Liau, J.Y.; Lee, C.H.; Jeng, Y.M. Lymphoepithelioma-like intrahepatic cholangiocarcinoma is a distinct entity with frequent pTERT/TP53 mutations and comprises 2 subgroups based on Epstein-Barr virus infection. Am. J. Surg. Pathol. 2021, 45, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Khandakar, B.; Liu, J.R.; Thung, S.; Li, Y.; Rhee, H.; Kagen, A.C.; Sun-Wing Tong, T.; Nyun Park, Y.; Theise, N.; Oi-Lin Ng, I. Lymphoepithelioma-like neoplasm of the biliary tract with ‘probable low malignant potential’. Histopathology 2022, 80, 720–728. [Google Scholar] [CrossRef]
- Nakajima, T.; Tajima, Y.; Sugano, I.; Nagao, K.; Kondo, Y.; Wada, K. Intrahepatic cholangiocarcinoma with sarcomatous change. Clinicopathologic and immunohistochemical evaluation of seven cases. Cancer 1993, 72, 1872–1877. [Google Scholar] [CrossRef]
- Kaibori, M.; Kawaguchi, Y.; Yokoigawa, N.; Yanagida, H.; Takai, S.; Kwon, A.H.; Uemura, Y.; Kamiyama, Y. Intrahepatic sarcomatoid cholangiocarcinoma. J. Gastroenterol. 2003, 38, 1097–1101. [Google Scholar] [CrossRef]
- Kim, D.K.; Kim, B.R.; Jeong, J.S.; Baek, Y.H. Analysis of intrahepatic sarcomatoid cholangiocarcinoma: Experience from 11 cases within 17 years. World J. Gastroenterol. 2019, 25, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Yoshuantari, N.; Jeng, Y.M.; Liau, J.Y.; Lee, C.H.; Tsai, J.H. Hepatic sarcomatoid carcinoma is an aggressive hepatic neoplasm sharing common molecular features with its conventional carcinomatous counterparts. Mod. Pathol. 2023, 36, 100042. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Kim, H.; Park, Y.N. Sarcomatoid cholangiocarcinoma with osteoclast-like giant cells associated with hepatolithiasis: A case report. Clin. Mol. Hepatol. 2015, 21, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Sintra, S.; Costa, R.; Filipe, C.; Simão, A. Intrahepatic sarcomatoid cholangiocarcinoma. BMJ Case Rep. 2018, 2018, bcr-2018-225017. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.K.; Wu, R.C.; Hung, C.F.; Lee, C.S. Intrahepatic sarcomatoid cholangiocarcinoma: Clinical analysis of seven cases during a 15-year period. Chang Gung Med. J. 2008, 31, 599–605. [Google Scholar] [PubMed]
- Xie, X.; Lai, N.; Yang, Y.; Zhang, J.; Qin, J.; Sheng, X. Pathologic features and clinical treatment of sarcomatoid intrahepatic cholangiocarcinoma. Intractable Rare Dis. Res. 2023, 12, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Zaher, E.A.; Patel, P.; Gotimukul, A.; Sqour, H. Sarcomatoid intrahepatic cholangiocarcinoma: A rare and aggressive primary liver cancer. Cureus 2023, 15, e39520. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.T.; Saka, B.; Zen, Y.; Pambuccian, S.; Bacchi, C.; Adsay, V. Tubulocystic carcinoma of bile ducts: A hitherto unrecognized and diagnostically challenging entity often mistaken as a benign lesion; clinicopathologic analysis of 6 cases. Mod. Pathol. 2013, 26, 404A. [Google Scholar]
- Amin, M.B.; MacLennan, G.T.; Gupta, R.; Grignon, D.; Paraf, F.; Vieillefond, A.; Paner, G.P.; Stovsky, M.; Young, A.N.; Srigley, J.R.; et al. Tubulocystic carcinoma of the kidney: Clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am. J. Surg. Pathol. 2009, 33, 384–392. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakamoto, Y.; Noguchi, H.; Yamada, S.; Hirata, K. Tubulocystic carcinoma of the bile duct. Case Reports Hepatol. 2018, 2018, 2304610. [Google Scholar] [CrossRef]
- Argani, P.; Palsgrove, D.N.; Anders, R.A.; Smith, S.C.; Saoud, C.; Kwon, R.; Voltaggio, L.; Assarzadegan, N.; Oshima, K.; Rooper, L.; et al. A novel NIPBL-NACC1 gene fusion is characteristic of the cholangioblastic variant of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 2021, 45, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Bakhshwin, A.; Lai, K.K.; Ammoun, A.; Friedman, K.; El Hag, M. Inhibin-positive “cholangioblastic” variant of intrahepatic cholangiocarcinoma: Report of 3 new patients with review of the literature. Int. J. Surg Pathol. 2023, 31, 10668969231157775. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, K.; Bacani, J.; Fung, C.; Canterbury, L.A. A cholangioblastic variant of cholangiocarcinoma. ACG. Case Rep. J. 2022, 9, e00746. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, E.; Hytiroglou, P.; Sikas, N.; Soultoyannis, I.; Goodman, Z.D. Hepatic adenocarcinoma expressing inhibin in a young patient on oral contraceptives. Virchows Arch. 2005, 446, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.W.; Joseph, N.M.; Srivastava, A.; Saunders, T.A.; Jain, D.; Rank, J.; Feely, M.; Zarrinpar, A.; Al Diffalha, S.; Shyn, P.B.; et al. Inhibin-positive hepatic carcinoma: Proposal for a solid-tubulocystic variant of intrahepatic cholangiocarcinoma. Hum. Pathol. 2021, 116, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Howard, L.N.; Alonsozana, E.; Sill, D.; Bose, D.; Lai, J. Molecular characteristics and immunogenomic profiling of cholangioblastic variant of intrahepatic cholangiocarcinoma in a 68-year-old patient. Anticancer Res. 2022, 42, 5475–5478. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M.S. Benign and malignant biliary tumors. In Biopsy Interpretation of the Liver, 4th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2020; pp. 815–817. ISBN 978-1-975157-29-6. [Google Scholar]
- Braxton, D.R.; Saxe, D.; Damjanov, N.; Stashek, K.; Shroff, S.; Morrissette, J.D.; Tondon, R.; Furth, E.E. Molecular and cytogenomic profiling of hepatic adenocarcinoma expressing inhibin A, a mimicker of neuroendocrine tumors: Proposal to reclassify as “cholangioblastic variant of intrahepatic cholangiocarcinoma”. Hum. Pathol. 2017, 62, 232–241. [Google Scholar] [CrossRef]
- Gushima, R.; Narita, R.; Shono, T.; Naoe, H.; Yao, T.; Sasaki, Y. Esophageal adenocarcinoma with enteroblastic differentiation arising in ectopic gastric mucosa in the cervical esophagus: A case report and literature review. J. Gastrointestin. Liver Dis. 2017, 26, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, R.; Wang, S.; Zhang, G. Extra-hepatic hepatoid carcinomas in female reproductive system: Three case-reports with a literature review. Cancer Manag. Res. 2021, 13, 1625–1636. [Google Scholar] [CrossRef]
- Muroyama, Y.; Tamiya, H.; Tanaka, G.; Tanaka, W.; Huang, A.C.; Oldridge, D.A.; Matsusaka, K.; Takazawa, Y.; Jo, T.; Ushiku, T.; et al. Alpha-fetoprotein-producing lung hepatoid adenocarcinoma with brain metastasis treated with S-1. Case Rep. Oncol. 2020, 13, 1552–1559. [Google Scholar] [CrossRef]
- Shiratori, Y.; Suzuki, K.; Ikeya, T. Colonic clear cell adenocarcinoma with enteroblastic differentiation. Clin. J. Gastroenterol. 2020, 13, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Fu, Y.; Sun, Q.; Geng, P.; Zheng, Z.; Pu, X.; Shi, J.; Fan, X. Integrated clinicopathological and immunohistochemical analysis of gastric adenocarcinoma with hepatoid differentiation: An exploration of histogenesis, molecular characteristics, and prognostic markers. Hum. Pathol. 2021, 115, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Yao, T.; Mitomi, H.; Morimoto, T.; Ueyama, H.; Matsumoto, K.; Saito, T.; Osada, T.; Nagahara, A.; Watanabe, S. Clinicopathologic and immunohistochemical characteristics of gastric adenocarcinoma with enteroblastic differentiation: A study of 29 cases. Gastric Cancer 2016, 19, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Canh, H.; Takahashi, K.; Yamamura, M.; Li, Z.; Sato, Y.; Yoshimura, K.; Kozaka, K.; Tanaka, M.; Nakanuma, Y.; Harada, K. Diversity in cell differentiation, histology, phenotype and vasculature of mass-forming intrahepatic cholangiocarcinomas. Histopathology 2021, 79, 731–750. [Google Scholar] [CrossRef]
- Okura, K.; Esaki, M.; Nara, S.; Ban, D.; Takamoto, T.; Shimada, K.; Hiraoka, N. Hepatoid carcinoma and related entities of the extrahepatic bile duct: A clinicopathological study of four cases. Pathol. Int. 2022, 72, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, R.T.; Mafficini, A.; Sciammarella, C.; Cantù, C.; Rusev, B.C.; Piredda, M.L.; Antonello, D.; Grimaldi, S.; Bonizzato, G.; Sperandio, N.; et al. Genomic characterization of hepatoid tumors: Context matters. Hum. Pathol. 2021, 118, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Zhou, Y.; Wang, Y.; Yuan, J.; Ma, X. Hepatoid adenocarcinoma of the stomach: Current perspectives and new developments. Front. Oncol. 2021, 11, 633916. [Google Scholar] [CrossRef] [PubMed]
- Ushiku, T.; Shinozaki, A.; Shibahara, J.; Iwasaki, Y.; Tateishi, Y.; Funata, N.; Fukayama, M. SALL4 represents fetal gut differentiation of gastric cancer, and is diagnostically useful in distinguishing hepatoid gastric carcinoma from hepatocellular carcinoma. Am. J. Surg. Pathol. 2010, 34, 533–540. [Google Scholar] [CrossRef]
- Liu, T.C.; Vachharajani, N.; Chapman, W.C.; Brunt, E.M. SALL4 immunoreactivity predicts prognosis in Western hepatocellular carcinoma patients but is a rare event: A study of 236 cases. Am. J. Surg. Pathol. 2014, 38, 966–972. [Google Scholar] [CrossRef]
- Arif, D.; Mettler, T.; Adeyi, O.A. Mimics of hepatocellular carcinoma: A review and an approach to avoiding histopathological diagnostic missteps. Hum. Pathol. 2021, 112, 116–127. [Google Scholar] [CrossRef]
- Emoto, K.; Eguchi, T.; Tan, K.S.; Takahashi, Y.; Aly, R.G.; Rekhtman, N.; Travis, W.D.; Adusumilli, P.S. Expansion of the concept of micropapillary adenocarcinoma to include a newly recognized filigree pattern as well as the classical pattern based on 1468 Stage I lung adenocarcinomas. J. Thorac. Oncol. 2019, 14, 1948–1961. [Google Scholar] [CrossRef]
- Zinnall, U.; Weyerer, V.; Compérat, E.; Camparo, P.; Gaisa, N.T.; Knuechel-Clarke, R.; Perren, A.; Lugli, A.; Toma, M.; Baretton, G.; et al. Micropapillary urothelial carcinoma: Evaluation of HER2 status and immunohistochemical characterization of the molecular subtype. Hum. Pathol. 2018, 80, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.M.; Bleiweiss, I.J. Invasive micropapillary carcinoma of the breast: Eighty cases of an underrecognized entity. Hum. Pathol. 2001, 32, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Konishi, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Kojima, M.; Kinoshita, T. Invasive micropapillary carcinoma of the ampulla of Vater with extensive lymph node metastasis: Report of a case. Surg. Today 2010, 40, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Khayyata, S.; Basturk, O.; Adsay, N.V. Invasive micropapillary carcinomas of the ampullo-pancreatobiliary region and their association with tumor-infiltrating neutrophils. Mod. Pathol. 2005, 18, 1504–1511. [Google Scholar] [CrossRef]
- Agaimy, A.; Kaiser, A.; Becker, K.; Bräsen, J.H.; Wünsch, P.H.; Adsay, N.V.; Klöppel, G. Pancreatic-type acinar cell carcinoma of the liver: A clinicopathologic study of four patients. Mod. Pathol. 2011, 24, 1620–1626. [Google Scholar] [CrossRef]
- Hervieu, V.; Lombard-Bohas, C.; Dumortier, J.; Boillot, O.; Scoazec, J.Y. Primary acinar cell carcinoma of the liver. Virchows Arch. 2008, 452, 337–341. [Google Scholar] [CrossRef]
- Jordan, E.J.; Basturk, O.; Shia, J.; Klimstra, D.S.; Alago, W.; D’Angelica, M.I.; Abou-Alfa, G.K.; O’Reilly, E.M.; Lowery, M.A. Case report: Primary acinar cell carcinoma of the liver treated with multimodality therapy. J. Gastrointest. Oncol. 2017, 8, E65–E72. [Google Scholar] [CrossRef]
- Sun, Y.; Wasserman, P.G. Acinar cell carcinoma arising in the stomach: A case report with literature review. Hum. Pathol. 2004, 35, 263–265. [Google Scholar] [CrossRef]
- Chiaravalli, A.M.; Finzi, G.; Bertolini, V.; La Rosa, S.; Capella, C. Colonic carcinoma with a pancreatic acinar cell differentiation. A case report. Virchows Arch. 2009, 455, 527–531. [Google Scholar] [CrossRef]
- Kawakami, H.; Kuwatani, M.; Onodera, M.; Hirano, S.; Kondo, S.; Nakanishi, Y.; Itoh, T.; Asaka, M. Primary acinar cell carcinoma of the ampulla of Vater. J. Gastroenterol. 2007, 42, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M.S. Liver. In Surgical Pathology Dissection: An Illustrated Guide, 2nd ed.; Westra, W.H., Hruban, R.H., Phelps, T.H., Isacson, C., Eds.; Springer: New York, NY, USA, 2003; pp. 76–81. ISBN 9-780387-95599. [Google Scholar]
- Suriawinata, A.A.; Thung, S.N. Liver Pathology: An Atlas and Concise Guide; Demos Medical Publishing: New York, NY, USA, 2011; pp. 1–15. ISBN 9-781933-864945. [Google Scholar]
- Westerhoff, M.; Lamps, L.W.; Kakar, S. Hepatectomy specimen handling. In Diagnostic Pathology: Hepatobiliary and Pancreas, 3rd ed.; Elsevier: Philadelphia, PA, USA, 2022; pp. 348–349. ISBN 978-0-323-77620-2. [Google Scholar]
- Torbenson, M. Masses of the liver. In Mills and Sternberg’s Diagnostic Surgical Pathology, 7th ed.; Longacre, T.A., Greenson, J.K., Hornick, J.L., Reuter, V.E., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2022; pp. 1890–1949. ISBN 978-1-975150-72-3. [Google Scholar]
- Lee, K.B. Histopathology of a benign bile duct lesion in the liver: Morphologic mimicker or precursor of intrahepatic cholangiocarcinoma. Clin. Mol. Hepatol. 2016, 22, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.; Borger, D.R.; Corless, C.; Hagen, C.; Iafrate, A.J.; Makhlouf, H.; Misdraji, J.; Sapp, H.; Tsui, W.M.; Wanless, I.R.; et al. Biliary adenofibroma of liver: Morphology, tumor genetics, and outcomes in 6 cases. Am. J. Surg. Pathol. 2017, 41, 499–505. [Google Scholar] [CrossRef]
- Tsokos, C.G.; Krings, G.; Yilmaz, F.; Ferrell, L.D.; Gill, R.M. Proliferative index facilitates distinction between benign biliary lesions and intrahepatic cholangiocarcinoma. Hum. Pathol. 2016, 57, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Assie, G.; Baudin, E.; Eisenhofer, G.; de la Fouchardiere, C.; Haak, H.R.; de Krijger, R.; Porpiglia, F.; Terzolo, M.; Berruti, A. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Loy, T.S.; Phillips, R.W.; Linder, C.L. A103 immunostaining in the diagnosis of adrenal cortical tumors: An immunohistochemical study of 316 cases. Arch. Pathol. Lab. Med. 2002, 126, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Asa, S.L.; Giordano, T.J.; Papotti, M.; Sasano, H.; Volante, M. Immunohistochemical biomarkers of adrenal cortical neoplasms. Endocr. Pathol. 2018, 29, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sarcognato, S.; Sacchi, D.; Fassan, M.; Fabris, L.; Cadamuro, M.; Zanus, G.; Cataldo, I.; Capelli, P.; Baciorri, F.; Cacciatore, M.; et al. Cholangiocarcinoma. Pathologica 2021, 113, 158–169. [Google Scholar] [CrossRef]
- Takahashi, Y.; Dungubat, E.; Kusano, H.; Ganbat, D.; Tomita, Y.; Odgerel, S.; Fukusato, T. Application of immunohistochemistry in the pathological diagnosis of liver tumors. Int. J. Mol. Sci. 2021, 22, 5780. [Google Scholar] [CrossRef]
- Nasir, A.; Lehrke, H.D.; Mounajjed, T.; Said, S.; Zhang, L.; Yasir, S.; Shah, S.S.; Chandan, V.S.; Smyrk, T.C.; Moreira, R.K.; et al. Albumin in situ hybridization can be positive in adenocarcinomas and other tumors from diverse sites. Am. J. Clin. Pathol. 2019, 152, 190–199. [Google Scholar] [CrossRef]
- Mocan, L.P.; Rusu, I.; Melincovici, C.S.; Boșca, B.A.; Mocan, T.; Crăciun, R.; Spârchez, Z.; Iacobescu, M.; Mihu, C.M. The role of immunohistochemistry in the differential diagnosis between intrahepatic cholangiocarcinoma, hepatocellular carcinoma and liver metastasis, as well as its prognostic value. Diagnostics 2023, 13, 1542. [Google Scholar] [CrossRef] [PubMed]
- Selves, J.; Long-Mira, E.; Mathieu, M.C.; Rochaix, P.; Ilié, M. Immunohistochemistry for diagnosis of metastatic carcinomas of unknown primary site. Cancers 2018, 10, 108. [Google Scholar] [CrossRef]
- Ai, D.; Yao, J.; Yang, F.; Huo, L.; Chen, H.; Lu, W.; Soto, L.M.S.; Jiang, M.; Raso, M.G.; Wang, S.; et al. TRPS1: A highly sensitive and specific marker for breast carcinoma, especially for triple-negative breast cancer. Mod. Pathol. 2021, 34, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Bachert, S.E.; Di, J.; Zhang, S.; Short, H.E.; Piecoro, D.W.; McDonald, R.J.; Myint, Z.W.; Hensley, P.J.; Allison, D.B. TRPS1 expression in primary and metastatic prostatic adenocarcinoma, muscle invasive bladder urothelial carcinoma, and breast carcinoma: Is TRPS1 truly specific and sensitive for a breast primary? Hum. Pathol. 2024, 143, 42–49. [Google Scholar] [CrossRef]
- Wang, H.L.; Kim, C.J.; Koo, J.; Zhou, W.; Choi, E.K.; Arcega, R.; Chen, Z.E.; Wang, H.; Zhang, L.; Lin, F. Practical immunohistochemistry in neoplastic pathology of the gastrointestinal tract, liver, biliary tract, and pancreas. Arch. Pathol. Lab. Med. 2017, 141, 1155–1180. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Chen, J.W.; He, X.S.; Zhang, H.Z.; Ling, Y.H.; Wen, J.H.; Deng, W.H.; Li, P.; Yun, J.P.; Xie, D.; et al. SATB2 is a promising biomarker for identifying a colorectal origin for liver metastatic adenocarcinomas. EBioMedicine 2018, 28, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, I. Testicular germ cell tumors: Serological and immunohistochemical diagnosis. Acta. Med. Acad. 2021, 50, 58–70. [Google Scholar] [CrossRef]
- Cao, D.; Guo, S.; Allan, R.W.; Molberg, K.H.; Peng, Y. SALL4 is a novel sensitive and specific marker of ovarian primitive germ cell tumors and is particularly useful in distinguishing yolk sac tumor from clear cell carcinoma. Am. J. Surg. Pathol. 2009, 33, 894–904. [Google Scholar] [CrossRef]
- Cheng, L.; Thomas, A.; Roth, L.M.; Zheng, W.; Michael, H.; Karim, F.W. OCT4: A novel biomarker for dysgerminoma of the ovary. Am. J. Surg. Pathol. 2004, 28, 1341–1346. [Google Scholar] [CrossRef]
- Xue, D.; Peng, Y.; Wang, F.; Allan, R.W.; Cao, D. RNA-binding protein LIN28 is a sensitive marker of ovarian primitive germ cell tumours. Histopathology 2011, 59, 452–459. [Google Scholar] [CrossRef]
- Vyas, M.; Jain, D. A practical diagnostic approach to hepatic masses. Indian J. Pathol. Microbiol. 2018, 61, 2–17. [Google Scholar] [CrossRef]
- Lennartz, M.; Gehrig, E.; Weidemann, S.; Gorbokon, N.; Menz, A.; Büscheck, F.; Hube-Magg, C.; Hinsch, A.; Reiswich, V.; Höflmayer, D.; et al. Large-scale tissue microarray evaluation corroborates high specificity of high-level arginase-1 immunostaining for hepatocellular carcinoma. Diagnostics 2021, 11, 2351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Schmidt, L.A.; Hatanaka, K.; Thomas, D.; Lagstein, A.; Myers, J.L. Evaluation of napsin A, TTF-1, p63, p40, and CK5/6 immunohistochemical stains in pulmonary neuroendocrine tumors. Am. J. Clin. Pathol. 2014, 142, 320–324. [Google Scholar] [CrossRef]
- Yatabe, Y.; Dacic, S.; Borczuk, A.C.; Warth, A.; Russell, P.A.; Lantuejoul, S.; Beasley, M.B.; Thunnissen, E.; Pelosi, G.; Rekhtman, N.; et al. Best practices recommendations for diagnostic immunohistochemistry in lung cancer. J. Thorac. Oncol. 2019, 14, 377–407. [Google Scholar] [CrossRef] [PubMed]
- Chapel, D.B.; Schulte, J.J.; Husain, A.N.; Krausz, T. Application of immunohistochemistry in diagnosis and management of malignant mesothelioma. Transl. Lung Cancer Res. 2020, 9, S3–S27. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Krausz, T.; Borczuk, A.; Cagle, P.T.; Chirieac, L.R.; Churg, A.; Galateau-Salle, F.; Gibbs, A.R.; et al. Guidelines for pathologic diagnosis of malignant mesothelioma: A consensus statement from the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2009, 133, 1317–1331. [Google Scholar] [CrossRef]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for pathologic diagnosis of malignant mesothelioma 2017 update of the consensus statement from the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef]
- Maleki, Z.; Nadella, A.; Nadella, M.; Patel, G.; Patel, S.; Kholová, I. INSM1, a novel biomarker for detection of neuroendocrine neoplasms: Cytopathologists’ view. Diagnostics 2021, 11, 2172. [Google Scholar] [CrossRef]
- Wang, X.Q.; Fu, J.; Zhang, Y.T.; Xu, Y. INSM1 expression in primary and metastatic neuroendocrine neoplasms at distinct locations. Pathol. Res. Pract. 2023, 253, 155067. [Google Scholar] [CrossRef]
- Heidarpour, M.; Tavanafar, Z. Diagnostic utility of PAX8 in differentiation of mullerian from non-mullerian tumors. Adv. Biomed. Res. 2014, 3, 96. [Google Scholar] [CrossRef]
- Köbel, M.; Rahimi, K.; Rambau, P.F.; Naugler, C.; Le Page, C.; Meunier, L.; de Ladurantaye, M.; Lee, S.; Leung, S.; Goode, E.L.; et al. An immunohistochemical algorithm for ovarian carcinoma typing. Int. J. Gynecol. Pathol. 2016, 35, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Aldyab, M.; El Jabbour, T.; Parilla, M.; Lee, H. Benign vs malignant pancreaticlesions: Molecular insights to an ongoing debate. World J. Gastrointest. Surg. 2021, 13, 406–418. [Google Scholar] [CrossRef]
- Schlitter, A.M.; Segler, A.; Steiger, K.; Michalski, C.W.; Jäger, C.; Konukiewitz, B.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci. Rep. 2017, 7, 41064. [Google Scholar] [CrossRef] [PubMed]
- Ritterhouse, L.L.; Wu, E.Y.; Kim, W.G.; Dillon, D.A.; Hirsch, M.S.; Sholl, L.M.; Agoston, A.T.; Setia, N.; Lauwers, G.Y.; Park, D.Y.; et al. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology 2019, 75, 546–551. [Google Scholar] [CrossRef]
- Gurel, B.; Ali, T.Z.; Montgomery, E.A.; Begum, S.; Hicks, J.; Goggins, M.; Eberhart, C.G.; Clark, D.P.; Bieberich, C.J.; Epstein, J.I.; et al. NKX3.1 as a marker of prostatic origin in metastatic tumors. Am. J. Surg. Pathol. 2010, 34, 1097–1105. [Google Scholar] [CrossRef]
- Beach, R.; Gown, A.M.; De Peralta-Venturina, M.N.; Folpe, A.L.; Yaziji, H.; Salles, P.G.; Grignon, D.J.; Fanger, G.R.; Amin, M.B. P504S immunohistochemical detection in 405 prostatic specimens including 376 18-gauge needle biopsies. Am. J. Surg. Pathol. 2002, 26, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Queisser, A.; Hagedorn, S.A.; Braun, M.; Vogel, W.; Duensing, S.; Perner, S. Comparison of different prostatic markers in lymph node and distant metastases of prostate cancer. Mod. Pathol. 2015, 28, 138–145. [Google Scholar] [CrossRef]
- Seipel, A.H.; Samaratunga, H.; Delahunt, B.; Wiklund, F.; Wiklund, P.; Lindberg, J.; Grönberg, H.; Egevad, L. Immunohistochemical profile of ductal adenocarcinoma of the prostate. Virchows Arch. 2014, 465, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Laury, A.R.; Perets, R.; Piao, H.; Krane, J.F.; Barletta, J.A.; French, C.; Chirieac, L.R.; Lis, R.; Loda, M.; Hornick, J.L.; et al. A comprehensive analysis of PAX8 expression in human epithelial tumors. Am. J. Surg. Pathol. 2011, 35, 816–826. [Google Scholar] [CrossRef]
- Li, H.; Jing, X.; Yu, J.; Liu, J.; Zhang, T.; Chen, S.; Zhang, X. A combination of cytokeratin 5/6, p63, p40 and MUC5AC are useful for distinguishing squamous cell carcinoma from adenocarcinoma of the cervix. Diagn. Pathol. 2020, 15, 104. [Google Scholar] [CrossRef]
- Marginean, E.C.; Gown, A.M.; Jain, D. Diagnostic approach to hepatic mass lesions and role of immunohistochemistry. Surg. Pathol. Clin. 2013, 6, 333–365. [Google Scholar] [CrossRef] [PubMed]
- Serag Eldien, M.M.; Abdou, A.G.; Elghrabawy, G.R.A.; Alhanafy, A.M.; Mahmoud, S.F. Stratification of urothelial bladder carcinoma depending on immunohistochemical expression of GATA3 and CK5/6. J. Immunoassay Immunochem. 2021, 42, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Mohanty, S.K.; Kunju, L.P.; Chang, E.; Chung, F.; Carvalho, J.C.; Paner, G.P.; Hansel, D.E.; Luthringer, D.J.; de Peralta-Ventrurina, M.N.; et al. Uroplakin II outperforms uroplakin III in diagnostically challenging settings. Histopathology 2014, 65, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, O.; Volmerig, J.; Dietel, M. Uroplakin III is a highly specific and moderately sensitive immunohistochemical marker for primary and metastatic urothelial carcinomas. Am. J. Clin. Pathol. 2000, 113, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, S.; Terris, B.; Rubbia-Brandt, L. Pathology of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Zhang, X.; Sheng, X.; Chang, X.Y.; Chen, J.; Chen, M.S.; Dong, H.; Duan, G.J.; Hu, H.P.; et al. Expert Consensus on Pathological Diagnosis of Intrahepatic Cholangiocarcinoma (2022 version). J. Clin. Transl. Hepatol. 2023, 11, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.Y.; Ma, J.Q.; Yun, J.P.; Chen, X.; Ling, Y.; Zhang, S.; Shi, J.Y.; Chang, Y.Q.; Ji, Y.; Wang, X.Y.; et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J. Hepatol. 2022, 76, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Personeni, N.; Aghemo, A.; Lleo, A. The immune milieu of cholangiocarcinoma: From molecular pathogenesis to precision medicine. J. Autoimmun. 2019, 100, 17–26. [Google Scholar] [CrossRef]
- Tomlinson, J.L.; Valle, J.W.; Ilyas, S.I. Immunobiology of cholangiocarcinoma. J. Hepatol. 2023, 79, 867–875. [Google Scholar] [CrossRef]
- Gupta, A.; Kurzrock, R.; Adashek, J.J. Evolution of the targeted therapy landscape for cholangiocarcinoma: Is cholangiocarcinoma the ‘NSCLC’ of GI oncology? Cancers 2023, 15, 1578. [Google Scholar] [CrossRef] [PubMed]
- Tavolari, S.; Brandi, G. Mutational landscape of cholangiocarcinoma according to different etiologies: A review. Cells 2023, 12, 1216. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2000 WHO Classification (3rd Edition) | 2010 WHO Classification (4th Edition) | 2019 WHO Classification (5th Edition) | |
|---|---|---|---|
| Tumor Category | Epithelial tumors, malignant | Epithelial tumors: biliary, malignant | Malignant biliary tumors |
| Tumor type and subtypes | Intrahepatic CCA (peripheral bile duct carcinoma) | Intrahepatic CCA | Conventional intrahepatic CCA Large duct intrahepatic CCA Small duct intrahepatic CCA |
| Cholangiolocarcinoma Intrahepatic CCA with ductal plate malformation pattern | |||
| Subtypes | Subtypes | Subtypes | |
| Cholangiolocellular carcinoma | Combined HCC-CCA with stem cell features, cholangiolocellular type (a) | ||
| Adenosquamous carcinoma | Adenosquamous carcinoma | Adenosquamous carcinoma | |
| Squamous cell carcinoma | Squamous cell carcinoma | Squamous cell carcinoma | |
| Mucinous carcinoma | Mucinous carcinoma | Mucinous carcinoma | |
| Signet-ring cell carcinoma | Signet-ring cell carcinoma | Signet-ring cell carcinoma | |
| Clear cell carcinoma | Clear cell carcinoma | Clear cell carcinoma | |
| Mucoepidermoid carcinoma | Mucoepidermoid carcinoma | Mucoepidermoid carcinoma | |
| Lymphoepithelioma-like carcinoma | Lymphoepithelioma-like carcinoma | Lymphoepithelioma-like carcinoma | |
| Sarcomatous intrahepatic CCA | Sarcomatous intrahepatic CCA | Sarcomatous intrahepatic CCA |
| Small-Duct Type | Large-Duct Type | |
|---|---|---|
| Main location | Peripheral hepatic parenchyma | Proximal to hepatic hilar regions |
| Risk factors | Hepatitis virus (HBV and HCV infection), alcoholic liver disease, metabolic syndrome, hemochromatosis, diabetes mellitus, obesity | Primary sclerosing cholangitis, hepatolithiasis, liver fluke infection |
| Precursors | Unknown | BilIN, IPNB, ITPN |
| Goss features | MF pattern | PI pattern, PI + MF pattern |
| Origins of cells | Small bile ducts and bile ductules, hepatic progenitor cells? | Intrahepatic large bile ducts, peribiliary glands |
| Histology | Small-ductal components: tubular pattern with low columnar to cuboidal cells and desmoplastic reaction Ductular components, cuboidal epithelia showing ductular or cord-like pattern with slit-like lumen and desmoplastic reaction | Ductal or tubular pattern with columnar to cuboidal epithelium, with desmoplastic reaction |
| Mucin production | Non–mucin-secreting glands | Mucin-secreting glands |
| Perineural/lymphatic invasion | Can be present | Common |
| Tumor border | Expansile or pushing, rarely infiltrative | Infiltrative |
| Molecular features | BAP1, IDH1/2 mutations, FGFR2 fusions, SMAD4, BAP1, BRAF, ARIDA1A, KRAS, TP53, SMAD4 mutations | KRAS mutations, TP53 mutations, SMAD4 mutations, MDM2 amplification |
| Immunohistochemical features | ||
| Common markers | EMA (MUC1), cytokeratin 7, cytokeratin 19 | EMA (MUC1), cytokeratin 7, cytokeratin 19 |
| Characteristic markers | CD56 (NCAM), C-reactive protein, N-cadherin, BAP1 (loss) | MUC5AC, MUC6, S100P, TFF1, AGR2, MMP7, SMAD4 (loss) |
| Similar to | Adenocarcinoma component of combined HCC-CCA | Perihilar CCA |
| Prognosis | Favorable (5-year survival 35–40%) | Poor (5-year survival 20–25%) |
| Subtype | Relative Frequency | Clinical Features | Pathological Features | Molecular Features | Prognosis (b) | References |
|---|---|---|---|---|---|---|
| Cholangiolocarcinoma | <5% | Share clinical and imaging features with both HCC and ICCA | >80% of the tumor composed of a ductular configuration, small cuboidal cells with round to oval nuclei with fine chromatin and scant cytoplasm, hyalinized fibrotic stroma | No distinct findings to date | Better | [82,83,84] |
| ICCA with ductal plate malformation pattern | <5% | 60% of patients have a history of chronic liver disease | >50% of the tumor shows tumor structures resembling those of ductal plate malformation | FGFR2 and PTPRT are most frequently mutated | Similar | [70,87,88] |
| Adenosquamous carcinoma | 2–3% | Associated with hepatolithiasis, hepatic cysts | Tumor composed of both glandular and squamous cell differentiation | No distinct findings to date | Worse | [91,92,93] |
| Squamous cell carcinoma | <1% | Associated with hepatic cyst, hepatolithiasis, hepatic teratoma | Entire tumor shows squamous differentiation | No distinct findings to date | Worse | [94,95,96,97,98,99,100,101] |
| Mucinous carcinoma | <1% | Unusual complication of hepatolithiasis and recurrent pyogenic cholangitis | >50% of the tumor composed of extracellular mucin pools and clusters of tumor cells; BilIN and IPNB can progress to mucinous carcinoma | Mucin synthesis by MUC4 and MUC16 is elevated via the up-regulated expression of mesothelin; transcription factor ONECUT3 | Better | [100,101,102,103,104,105,106] |
| Signet ring cell carcinoma | <1% | No distinct findings to date | >50% of the tumor composed of signet ring cells | No distinct findings to date | Unclear | [110,111,112,113,114] |
| Clear cell carcinoma | <1% | No distinct findings to date | >50% of the tumor composed of clear cells | No distinct findings to date | Better | [115,116,117,120] |
| Mucoepidermoid carcinoma | <1% | No distinct findings to date | Tumor composed of mucous, intermediate, and epidermoid cells | Most cases lack CRTC1::MAML2 fusion | Worse | [122,123,124,125,126] |
| Lymphoepithelioma-like carcinoma | <5% | Associated with Epstein–Barr virus | Tumor cells arranged in sheets and abortive glands with dense lymphoplasmacytic infiltrate | Frequent mutations in pTERT and TP53 | Better | [129,130,131,132,133,134,135,136,137] |
| Sarcomatous ICCA | <5% | Associated with chronic hepatitis B and C, hepatolithiasis | Spindle or pleomorphic giant tumor cells with focal adenocarcinoma component | Rare mutations in pTERT and TP53 mutations | Worse | [138,139,140,141,142,143,144] |
| Subtype | Relative Frequency | Clinical Features | Pathological Features | Molecular Features | Prognosis (b) | References |
|---|---|---|---|---|---|---|
| Tubulocystic carcinoma of the bile duct | <1% | No distinct findings to date | Cystically dilated tubules with intracystic papillary growth, grossly sponge-like appearance | No distinct findings to date | Unclear | [147,148,149] |
| Cholangioblastic (solid-tubulocystic or thyroid follicle-like) cholangiocarcinoma | <1% | Younger women (average age, approximately 40 years) | Wide range of morphology, including solid, trabecular, microcystic, follicular, blastemal-like areas | NIPBL::NACC1 fusion | Unclear | [150,151,152,153,154,155,156,157] |
| Enteroblastic cholangiocarcinoma | <1% | Extremely rare, especially extrahepatic bile duct | Polygonal cells with tubular and papillary growth or columnar with clear cytoplasm (fetal gut-like) | Loss of CDKN2A and loss of chromosome 18 | Worse | [158,159,160,161,162,163,164,165] |
| Micropapillary carcinoma | <1% | No distinct findings to date | Micropapillary clusters without fibrovascular cores | No distinct findings to date | Worse | [171,172,173] |
| Acinar cell carcinoma | <1% | No distinct findings to date | Uniform round nuclei with moderate amount of granular, eosinophilic to amphophilic cytoplasm containing zymogen granules | No distinct findings to date | Better | [176,177,178] |
| Tumor Origin | Markers | NOTE | References |
|---|---|---|---|
| Adrenocortical | SF1, inhibin, Mart-1/Melan-A, synaptophysin, calretinin | SF1 is the most reliable biomarker with which to confirm the cortical origin | [189,190,191] |
| Bile duct | CK7, CK19, CA19-9, CEA | CK19 and CA19-9 show the highest sensitivity in ICCA | [192,193,194,195] |
| Breast | ER, PR, GCDFP-15, mammaglobin, GATA3, TRPS1 | TRPS1 is a highly sensitive and specific marker for breast carcinoma; however, it causes significant staining in urinary bladder and prostate cancer | [192,196,197,198] |
| Colorectal | CK20, MUC2, CDX2, SATB2 | An antibody panel of CK7, CK20, CDX2, SATB2, and MUC2 can aid in the distinction between ICCA and metastatic colorectal adenocarcinomas | [192,199,200] |
| Germ cell | PLAP, OCT4, hCG, CD30, SALL4, LIN28, CD117, D2-40, SOX2, AFP, glypican-3 | PLAP, OCT4, hCG, and CD30 are commonly used markers for detecting germ cell tumors | [201,202,203,204,205] |
| Hepatocellular | Hep Par-1, arginase-1, CD10, polyclonal CEA, AFP | Arginase-1 is a highly sensitive and specific marker for HCC and is better than Hep Par-1 in poorly differentiated HCC | [196,199,206] |
| Lung | CK7, TTF-1, Napsin A | TTF-1 is widely used as a specific marker for pulmonary adenocarcinoma but can be expressed in neuroendocrine tumors, papillary thyroid carcinoma, and some female genital tract carcinomas | [196,199,207,208] |
| Mesothelial | Calretinin, D2-40 (podoplanin), WT1, CK5/6 | It is advisable to use panels of positive mesothelial markers (three or four) and negative antibodies | [196,209,210,211] |
| Neuroendocrine | Chromogranin A, synaptophysin, CD56, INSM1 | Chromogranin A is more specific than synaptophysin; INSM1 is a recently discovered, useful neuroendocrine marker in primary and metastatic NETs | [192,199,212,213] |
| Ovarian | PAX8, WT1, Napsin A, CA125, PR, ER | A panel consisting of PAX-8, WT1, and CA125 is useful for the diagnosis of primary ovarian carcinoma | [214,215] |
| Pancreatic duct | CK7, CK19, SMAD4, p16 | CK7 and CK19 are usually positive in ICCA and PDA; loss of SMAD4 expression is more common in PDA than in ICCA | [192,199,216,217,218] |
| Prostate | PSA, PSAP, PSMA, NKX3.1, P504S (AMACR), | PSA is a specific marker for prostatic carcinoma but approximately 10% of high-grade prostatic carcinoma are negative for PSA; other prostatic-specific markers such as NKX3.1 are useful for confirming the diagnosis | [196,219,220,221,222] |
| Renal | CD10, PAX2, PAX8, vimentin, CAIX, RCC marker | PAX-8 is expressed in a wide range of tumors and must be used as a part of diagnostic panels, including CAIX and PAX2 | [196,205,223] |
| Squamous | CK5/6, p40, p63 | CK5/6, p40, and p63 are useful for confirming squamous cell carcinoma | [224,225] |
| Urothelial | GATA3, p63, uroplakin, CK5/6 | GATA3, p63, and uroplakin are most useful for confirming metastatic urothelial carcinoma | [196,226,227,228] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Thung, S.N. Recent Advances in Pathology of Intrahepatic Cholangiocarcinoma. Cancers 2024, 16, 1537. https://doi.org/10.3390/cancers16081537
Choi JH, Thung SN. Recent Advances in Pathology of Intrahepatic Cholangiocarcinoma. Cancers. 2024; 16(8):1537. https://doi.org/10.3390/cancers16081537
Chicago/Turabian StyleChoi, Joon Hyuk, and Swan N. Thung. 2024. "Recent Advances in Pathology of Intrahepatic Cholangiocarcinoma" Cancers 16, no. 8: 1537. https://doi.org/10.3390/cancers16081537
APA StyleChoi, J. H., & Thung, S. N. (2024). Recent Advances in Pathology of Intrahepatic Cholangiocarcinoma. Cancers, 16(8), 1537. https://doi.org/10.3390/cancers16081537