
Understanding the Role of Connexins in Hepatocellular Carcinoma: Molecular and Prognostic Implications

 ,
,  , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Connexin Structure, Regulation, and Involvement in Tumorigenic Processes
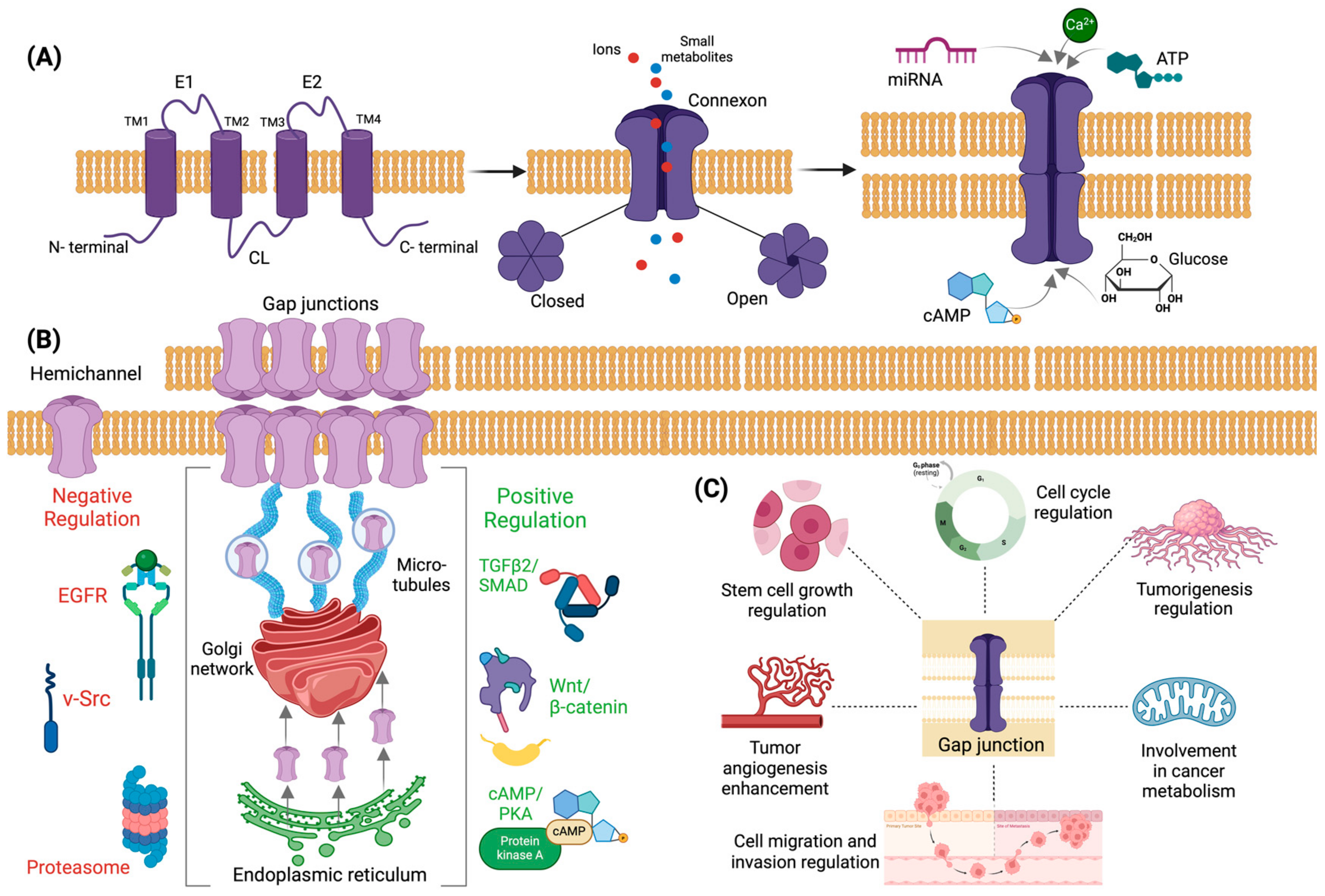
2.1. Structure and Formation of Gap Junctions and Connexins
2.2. Regulation of Connexins Expression
2.3. Connexins in Neoplastic Processes
3. Defining Connexins’ Expression and Functionality as Potential Prognostic and Therapeutic Markers in HCC
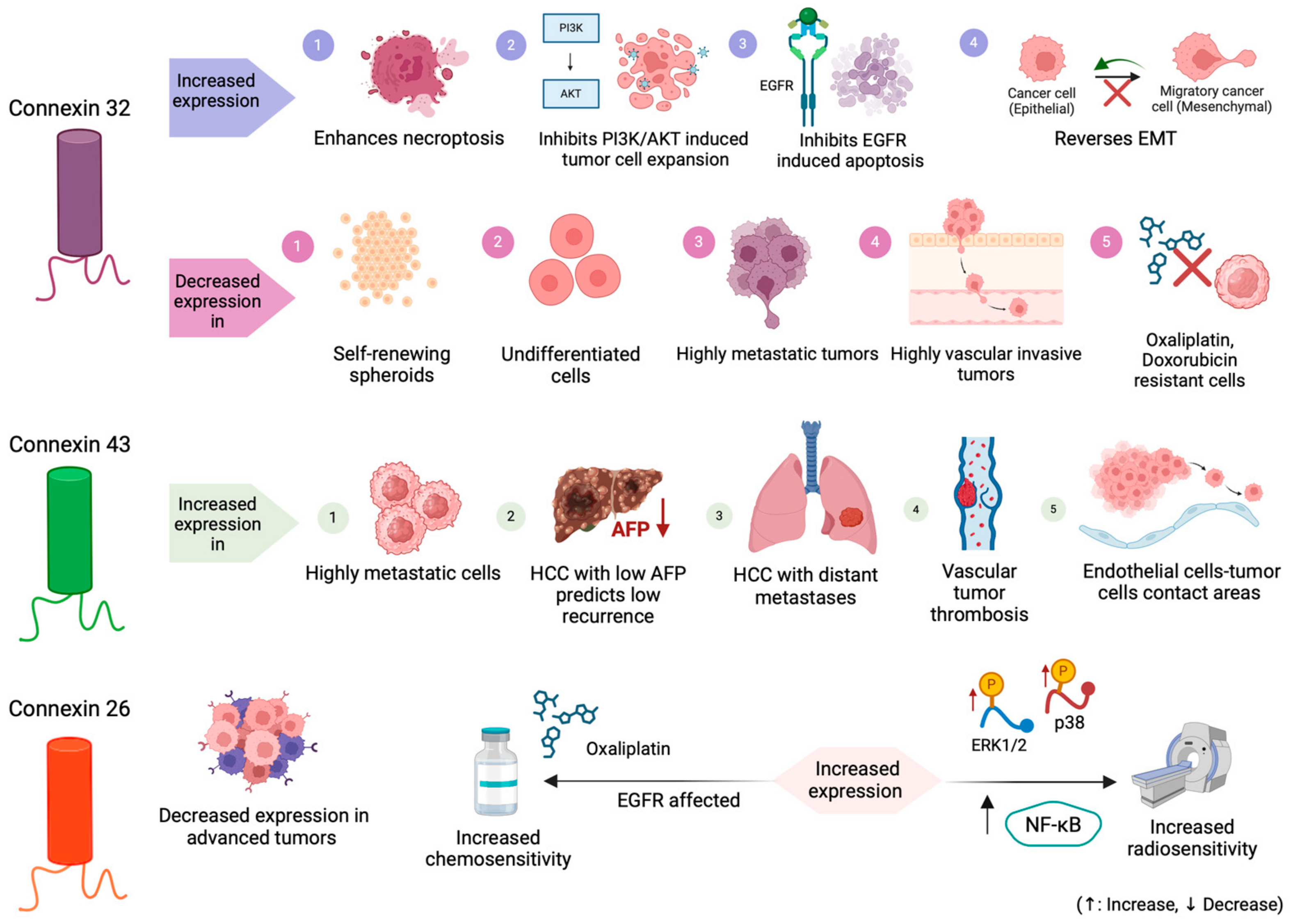
3.1. Connexin 32 (Cx32)
3.1.1. Connexin 32 Expression
3.1.2. The Connexin 32 Role in Tumorigenesis and Metastasis in HCC
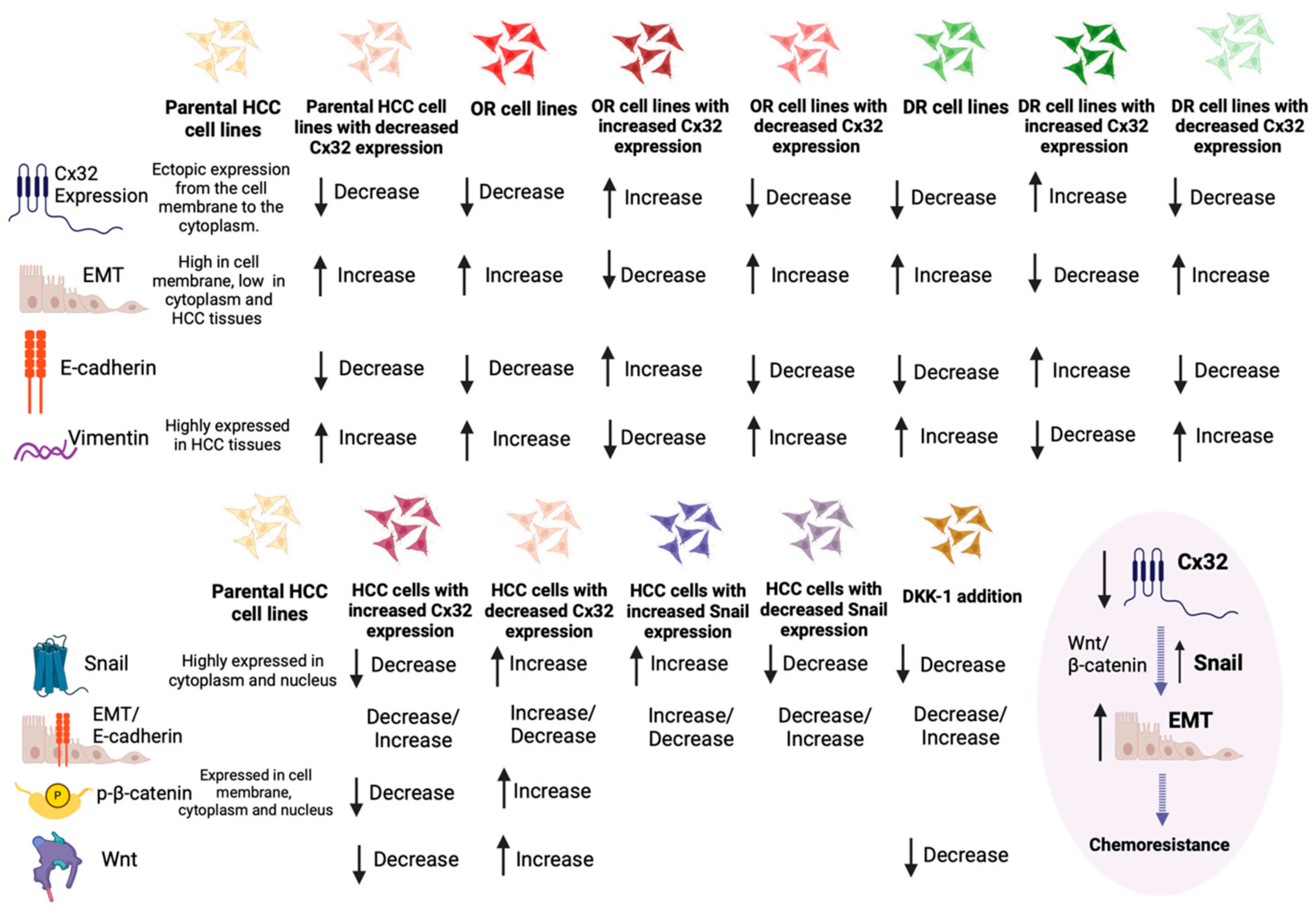
3.1.3. Exploring the Interplay between Connexin 32, EMT Signaling, and Chemoresistance in HCC
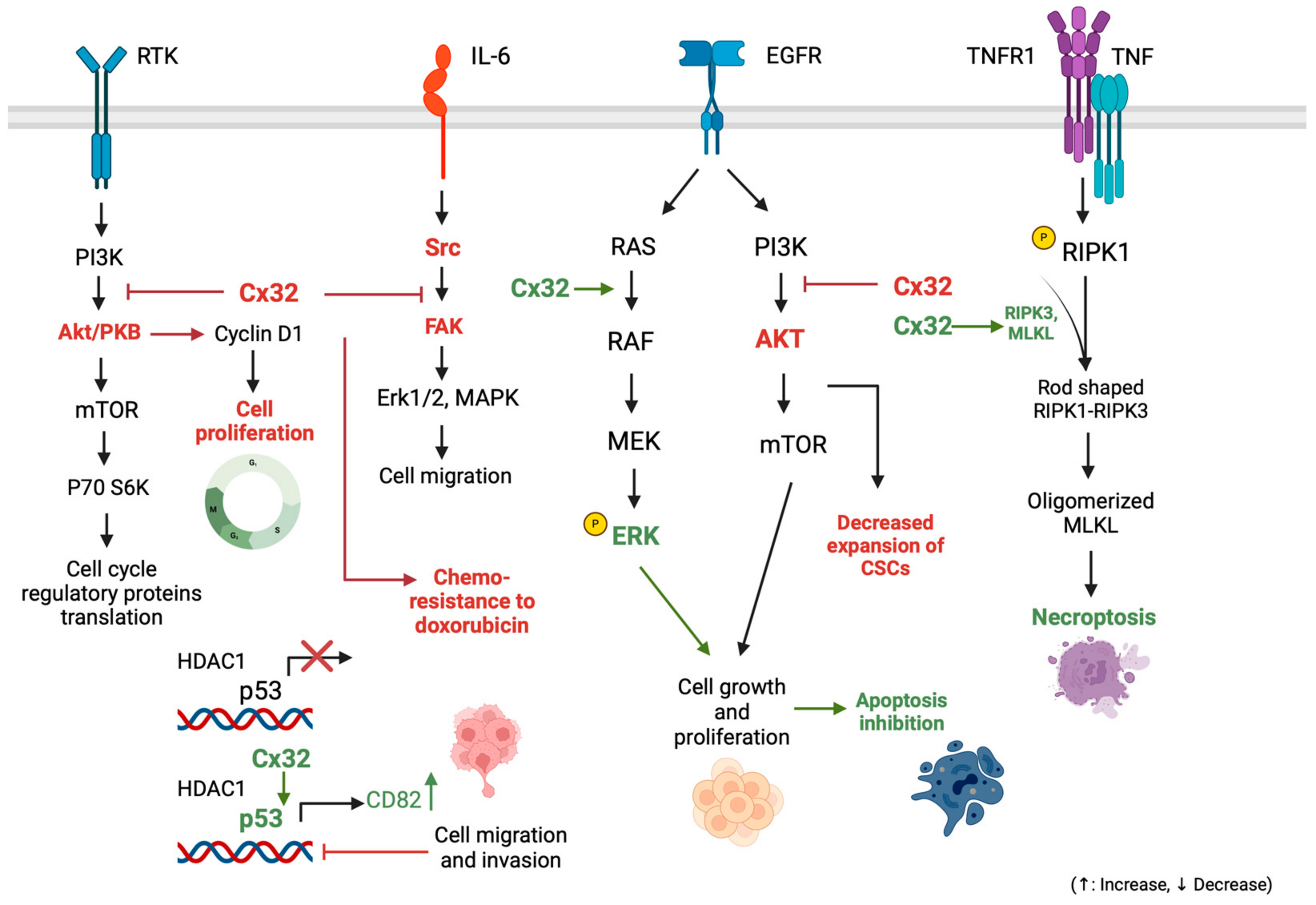
3.1.4. Connexin 32: Implications in Cell Survival-Proliferation Pathways and Other Tumorigenic Mechanisms
3.1.5. Exploring Connexin 32’s Role in Modulating Apoptosis and Necroptosis Pathways in HCC
3.1.6. Connexin 32 and Pre-Cancerous HCC-Related Conditions
3.2. Connexin 43 (Cx43)
3.3. Connexin 26 (Cx26)
4. Discussion
5. Conclusions—Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Liver Biol. Pathobiol. 2020, 47, 758–772. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127 (Suppl. S1), S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Güthle, M.; Dollinger, M.M. Epidemiology and risk factors of hepatocellular carcinoma. Radiologe 2014, 54, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinogenesis 2017, 16, 1–8. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Yanguas, S.C.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; da Silva, T.C.; Pereira, I.V.A.; Jaeschke, H.; Cogliati, B.; et al. Structure, Regulation and Function of Gap Junctions in Liver. Cell Commun. Adhes. 2016, 22, 29–37. [Google Scholar] [CrossRef]
- Centonze, L.; Di Sandro, S.; Lauterio, A.; De Carlis, R.; Frassoni, S.; Rampoldi, A.; Tuscano, B.; Bagnardi, V.; Vanzulli, A.; De Carlis, L. Surgical Resection vs. Percutaneous Ablation for Single Hepatocellular Carcinoma: Exploring the Impact of Li-RADS Classification on Oncological Outcomes. Cancers 2021, 13, 1671. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Guerra, M.; Hadjihambi, A.; Jalan, R. Gap junctions in liver disease: Implications for pathogenesis and therapy. J. Hepatol. 2019, 70, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Giaume, C. Bridging the gap to therapeutic strategies based on connexin/pannexin biology. J. Transl. Med. 2016, 14, 330. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Eugenín, E.A.; González, H.E.; Sánchez, H.A.; Brañes, M.C.; Sáez, J.C. Inflammatory conditions induce gap junctional communication between rat Kupffer cells both in vivo and in vitro. Cell. Immunol. 2007, 247, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wilgenbus, K.K.; Kirkpatrick, C.J.; Knuechel, R.; Willecke, K.; Traub, O. Expression of Cx26, Cx32 AND Cx43 gap junction proteins in normal and neoplastic human tissues. Int. J. Cancer 1992, 51, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Martinez, E.; Landazuri-Navas, S.; Vilchez, E.; Cantu-Hernandez, R.; Mosquera-Moscoso, J.; Encalada, S.; Al Lami, Z.; Zevallos-Delgado, C.; Cinicola, J. Prognostic Scores and Survival Rates by Etiology of Hepatocellular Carcinoma: A Review. J. Clin. Med. Res. 2023, 15, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Li, Y.; Qian, J.; Ding, S.; Sun, M.; Tan, B.; Zhao, Y. Connexin26 Modulates the Radiosensitivity of Cutaneous Squamous Cell Carcinoma by Regulating the Activation of the MAPK/NF-κB Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 672571. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, J.; Zhang, N.; Zhao, Y.; Li, W.-Y.; Zhao, F.-Y.; Ou, Y.-R.; Qin, S.-K.; Wu, Q. Impaired gap junctions in human hepatocellular carcinoma limit intrinsic oxaliplatin chemosensitivity: A key role of connexin 26. Int. J. Oncol. 2016, 48, 703–713. [Google Scholar] [CrossRef]
- Wilk, A.J.; Shalek, A.K.; Holmes, S.; Blish, C.A. Comparative analysis of cell–cell communication at single-cell resolution. Nat. Biotechnol. 2024, 42, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Xiang, J. Mechanisms of cellular communication through intercellular protein transfer. J. Cell. Mol. Med. 2011, 15, 1458–1473. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Sánchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, N.; Kalashnikova, M.; Belyavsky, A. Non-Classical Intercellular Communications: Basic Mechanisms and Roles in Biology and Medicine. Int. J. Mol. Sci. 2023, 24, 6455. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Richard, G.; White, T.W. Gap Junctions: Basic Structure and Function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zheng, M.; Zhou, X.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. The roles of connexins and gap junctions in the progression of cancer. Cell Commun. Signal. 2023, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Mroue, R.M.; El-Sabban, M.E.; Talhouk, R.S. Connexins: A myriad of functions extending beyond assembly of gap junction channels. Cell Commun. Signal. 2009, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, D.; Chen, X. Dynamic regulation of plasmodesmatal permeability and its application to horticultural research. Hortic. Res. 2019, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. An Update on Connexin Genes and their Nomenclature in Mouse and Man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.G. Connexins and pannexins: Coordinating cellular communication in the testis and epididymis. Spermatogenesis 2011, 1, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; de Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Cartenì, M.; Generoso, M.; et al. Zonulin Upregulation Is Associated with Increased Gut Permeability in Subjects With Type 1 Diabetes and Their Relatives. Diabetes 2006, 55, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Segretain, D.; Falk, M.M. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim. Biophys. Acta (BBA) Biomembr. 2004, 1662, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, W.; Wei, J.; Cui, Y.; Zhang, D.; Xie, J. Transforming growth factor-β1-induced N-cadherin drives cell–cell communication through connexin43 in osteoblast lineage. Int. J. Oral Sci. 2021, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Liu, Y.; Guo, D.; Kan, S.; Niu, Z.; Pu, X.; Bai, M.; Zhang, D.; Du, W.; Xie, J. TGF-β2 increases cell-cell communication in chondrocytes via p-Smad3 signalling. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2022, 1869, 119175. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.G.; Verlaan, I.; Moolenaar, W.H. Connexin-43 Interactions with ZO-1 and α- and β-tubulin. Cell Commun. Adhes. 2001, 8, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Macquart, C.; Jüttner, R.; Rodriguez, B.M.; Le Dour, C.; Lefebvre, F.; Chatzifrangkeskou, M.; Schmitt, A.; Gotthardt, M.; Bonne, G.; Muchir, A. Microtubule cytoskeleton regulates Connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. 2019, 28, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Thévenin, A.F.; Margraf, R.A.; Fisher, C.G.; Kells-Andrews, R.M.; Falk, M.M. Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 2017, 28, 3595–3608. [Google Scholar] [CrossRef] [PubMed]
- Dukic, A.R.; Gerbaud, P.; Guibourdenche, J.; Thiede, B.; Taskén, K.; Pidoux, G. Ezrin-anchored PKA phosphorylates serine 369 and 373 on connexin 43 to enhance gap junction assembly, communication, and cell fusion. Biochem. J. 2018, 475, 455–476. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.; Hartfield, E.M.; A Crompton, L.; Badger, J.L.; Glover, C.P.; Kelly, C.M.; E Rosser, A.; Uney, J.B.; A Caldwell, M. Cross-regulation of Connexin43 and β-catenin influences differentiation of human neural progenitor cells. Cell Death Dis. 2014, 5, e1017. [Google Scholar] [CrossRef] [PubMed]
- Kopanic, J.L.; Schlingmann, B.; Koval, M.; Lau, A.F.; Sorgen, P.L.; Su, V.F. Degradation of gap junction connexins is regulated by the interaction with Cx43-interacting protein of 75 kDa (CIP75). Biochem. J. 2015, 466, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M. Autophagy: A pathway that contributes to connexin degradation. J. Cell Sci. 2011, 124, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Carette, D.; Gilleron, J.; Denizot, J.; Grant, K.; Pointis, G.; Segretain, D. New cellular mechanisms of gap junction degradation and recycling. Biol. Cell 2015, 107, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Nimlamool, W.; Andrews, R.M.K.; Falk, M.M. Connexin43 phosphorylation by PKC and MAPK signals VEGF-mediated gap junction internalization. Mol. Biol. Cell 2015, 26, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-P.; Zhou, Y.; Hou, L.-X.; Zhu, X.-X.; Yi, W.; Yang, S.-M.; Lin, T.-Y.; Huang, J.-L.; Zhang, B.; Yin, X.-X. Cx43 deficiency confers EMT-mediated tamoxifen resistance to breast cancer via c-Src/PI3K/Akt pathway. Int. J. Biol. Sci. 2021, 17, 2380–2398. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, S.J.; Angenent, S.B.; Wang, Y.; Wu, L.F. On the spontaneous emergence of cell polarity. Nature 2008, 454, 886–889. [Google Scholar] [CrossRef]
- Bornens, M. Organelle positioning and cell polarity. Nat. Rev. Mol. Cell Biol. 2008, 9, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2012, 12, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, S.; Hung, H.; Chang, C.; Wu, I.; Huang, Y.; Lin, T.; Tsai, J.; Chen, A.; Kuo, F.; et al. Isolation and characterization of human gastric cell lines with stem cell phenotypes. J. Gastroenterol. Hepatol. 2007, 22, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [PubMed]
- Koffler, L.; Roshong, S.; Park, I.K.; Cesen-Cummings, K.; Thompson, D.C.; Dwyer-Nield, L.D.; Rice, P.; Mamay, C.; Malkinson, A.M.; Ruch, R.J. Growth inhibition in G1 and altered expression of cyclin D1 and p27kip-1after forced connexin expression in lung and liver carcinoma cells. J. Cell. Biochem. 2000, 79, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-L.; Huang, Y.-S.; Hosokawa, M.; Miyashita, K.; Hu, M.-L. Inhibition of proliferation of a hepatoma cell line by fucoxanthin in relation to cell cycle arrest and enhanced gap junctional intercellular communication. Chem. Interactions 2009, 182, 165–172. [Google Scholar] [CrossRef]
- Swietach, P.; Monterisi, S. A Barter Economy in Tumors: Exchanging Metabolites through Gap Junctions. Cancers 2019, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chen, C.; Li, Y.; Fu, X.; Xie, Y.; Li, Y.; Huang, Y. Cx31.1 acts as a tumour suppressor in non-small cell lung cancer (NSCLC) cell lines through inhibition of cell proliferation and metastasis. J. Cell. Mol. Med. 2012, 16, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yanguas, S.C.; Willebrords, J.; Weemhoff, J.L.; da Silva, T.C.; Decrock, E.; Lebofsky, M.; Pereira, I.V.A.; Leybaert, L.; Farhood, A.; et al. Connexin hemichannel inhibition reduces acetaminophen-induced liver injury in mice. Toxicol. Lett. 2017, 278, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yao, J.-H.; Du, Q.-Y.; Zhou, Y.-C.; Yao, T.-J.; Wu, Q.; Liu, J.; Ou, Y.-R. Connexin 32 downregulation is critical for chemoresistance in oxaliplatin-resistant HCC cells associated with EMT. Cancer Manag. Res. 2019, 11, 5133–5146. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Ono, T.; Yamanoi, A.; El-Assal, O.N.; Kohno, H.; Nagasue, N. Expression of gap junction protein connexin32 in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. J. Gastroenterol. 2004, 39, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.O.; Jondhale, S.; Chen, T.; Chipman, J.K. A quantitative inverse relationship between connexin32 expression and cell proliferation in a rat hepatoma cell line. Toxicology 2008, 253, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-D.; Ma, X.; Sui, Y.-F.; Wang, W.-L. Expression of gap junction genes connexin 32 and connexin 43 mRNAs and proteins, and their role in hepatocarcinogenesis. World J. Gastroenterol. 2002, 8, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Sheen, I.-S.; Jeng, K.-S.; Wang, P.-C.; Shih, S.-C.; Chang, W.-H.; Wang, H.-Y.; Chen, C.-C.; Shyung, L.-R. Are gap junction gene connexins 26, 32 and 43 of prognostic values in hepatocellular carcinoma? A prospective study. World J. Gastroenterol. 2004, 10, 2785–2790. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Omori, Y.; Nishikawa, Y.; Yoshioka, T.; Yamamoto, Y.; Enomoto, K. Cytoplasmic accumulation of connexin32 protein enhances motility and metastatic ability of human hepatoma cells in vitro and in vivo. Int. J. Cancer 2007, 121, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Omori, Y.; Li, Q.; Nishikawa, Y.; Yoshioka, T.; Yoshida, M.; Ishikawa, K.; Enomoto, K. Cytoplasmic accumulation of connexin32 expands cancer stem cell population in human HuH7 hepatoma cells by enhancing its self-renewal. Int. J. Cancer 2011, 128, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, N.; Zhu, J.; Hong, X.-T.; Liu, H.; Ou, Y.-R.; Su, F.; Wang, R.; Li, Y.-M.; Wu, Q. Downregulated connexin32 promotes EMT through the Wnt/β-catenin pathway by targeting Snail expression in hepatocellular carcinoma. Int. J. Oncol. 2017, 50, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Han, G.; Qi, B.; Wu, X. Cx32 reverses epithelial-mesenchymal transition in doxorubicin-resistant hepatocellular carcinoma. Oncol. Rep. 2017, 37, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zou, Q.; Wu, X.; Han, G.; Tong, X. Connexin 32 affects doxorubicin resistance in hepatocellular carcinoma cells mediated by Src/FAK signaling pathway. Biomed. Pharmacother. 2017, 95, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhao, W.; Wang, Y.; Xu, Y.; Xu, J.; Tang, K.; Zhang, S.; Yin, Z.; Wu, Q.; Wang, X. Connexin32 regulates hepatoma cell metastasis and proliferation via the p53 and Akt pathways. Oncotarget 2015, 6, 10116–10133. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, B.; Qi, B.; Jiang, G.; Qin, M.; Yu, M. Connexin32 regulates expansion of liver cancer stem cells via the PI3K/Akt signaling pathway. Oncol. Rep. 2022, 48, 166. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Wang, Q.; Guo, Y.; Ge, H.; Fu, Y.; Wang, X.; Tao, L. Cx32 exerts anti-apoptotic and pro-tumor effects via the epidermal growth factor receptor pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 145. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Peng, F.; Guo, Y.; Ge, H.; Cai, S.; Fan, L.; Peng, Y.; Wen, H.; Wang, Q.; Tao, L. Connexin32 activates necroptosis through Src-mediated inhibition of caspase 8 in hepatocellular carcinoma. Cancer Sci. 2021, 112, 3507–3519. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Naiki-Ito, A.; Naiki, T.; Suzuki, S.; Yamashita, Y.; Sato, S.; Sagawa, H.; Kato, A.; Kuno, T.; Takahashi, S. Connexin 32 dysfunction promotes ethanol-related hepatocarcinogenesis via activation of Dusp1-Erk axis. Oncotarget 2016, 7, 2009–2021. [Google Scholar] [CrossRef] [PubMed]
- Sagawa, H.; Naiki-Ito, A.; Kato, H.; Naiki, T.; Yamashita, Y.; Suzuki, S.; Sato, S.; Shiomi, K.; Kato, A.; Kuno, T.; et al. Connexin 32 and luteolin play protective roles in non-alcoholic steatohepatitis development and its related hepatocarcinogenesis in rats. Carcinogenesis 2015, 36, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Timmen, U.; Traub, O.; Dermietzel, R.; Rabes, H.; Willecke, K. Reduced number of gap junctions in rat hepatocarcinomas detected by monoclonal antibody. Carcinogenesis 1986, 7, 1475–1482. [Google Scholar] [CrossRef]
- Omori, Y.; Krutovskikh, V.; Mironov, N.; Tsuda, H.; Yamasaki, H. Cx32 gene mutation in a chemically induced rat liver tumour. Carcinogenesis 1996, 17, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, P.J.; Gartler, S.M.; Yoshida, A. Clonal origin of chronic myelocytic leukemia in man. Proc. Natl. Acad. Sci. USA 1967, 58, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Shimano, K.; Satake, M.; Okaya, A.; Kitanaka, J.; Kitanaka, N.; Takemura, M.; Sakagami, M.; Terada, N.; Tsujimura, T. Hepatic Oval Cells Have the Side Population Phenotype Defined by Expression of ATP-Binding Cassette Transporter ABCG2/BCRP1. Am. J. Pathol. 2003, 163, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Falciatori, I.; Borsellino, G.; Haliassos, N.; Boitani, C.; Corallini, S.; Battistini, L.; Bernardi, G.; Stefanini, M.; Vicini, E. Identification and enrichment of spermatogonial stem cells displaying side-population phenotype in immature mouse testis. FASEB J. 2004, 18, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- van Zijl, F.; Zulehner, G.; Petz, M.; Schneller, D.; Kornauth, C.; Hau, M.; Machat, G.; Grubinger, M.; Huber, H.; Mikulits, W.; et al. Epithelial–mesenchymal transition in hepatocellular carcinoma. Futur. Oncol. 2009, 5, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Brozovic, A. The relationship between platinum drug resistance and epithelial–mesenchymal transition. Arch. Toxicol. 2017, 91, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Hou, Y.; Yang, Q.; Chen, S.; Wang, X.; Wang, Z.; Yang, Y.; Chen, C.; Wang, Z.; et al. The PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal transition in gemcitabine resistance hepatoma cells. Oncotarget 2015, 6, 7000–7010. [Google Scholar] [CrossRef] [PubMed]
- Niknami, Z.; Muhammadnejad, A.; Ebrahimi, A.; Harsani, Z.; Shirkoohi, R. Significance of e-cadherin and vimentin as epithelial-mesenchymal transition markers in colorectal carcinoma prognosis. EXCLI J. 2020, 19, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2014, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-J.; Dai, Z.; Zhou, S.-L.; Hu, Z.-Q.; Chen, Q.; Zhao, Y.-M.; Shi, Y.-H.; Gao, Q.; Wu, W.-Z.; Qiu, S.-J.; et al. HNRNPAB Induces Epithelial–Mesenchymal Transition and Promotes Metastasis of Hepatocellular Carcinoma by Transcriptionally Activating SNAIL. Cancer Res. 2014, 74, 2750–2762. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-T.; Dai, Z.; Song, K.; Zhang, Z.-J.; Zhou, Z.-J.; Zhou, S.-L.; Zhao, Y.-M.; Xiao, Y.-S.; Sun, Q.-M.; Ding, Z.-B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [PubMed]
- van Veelen, W.; Le, N.H.; Helvensteijn, W.; Blonden, L.; Theeuwes, M.; Bakker, E.R.M.; Franken, P.F.; van Gurp, L.; Meijlink, F.; van der Valk, M.A.; et al. β-catenin tyrosine 654 phosphorylation increases Wnt signalling and intestinal tumorigenesis. Gut 2011, 60, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J. Gastroenterol. 2009, 15, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Matsubara, K.; Qian, G.-S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, L.G.; Bermejo, F.J.J.; Boaru, D.L.; De Castro-Martinez, P.; De Leon-Oliva, D.; Fraile-Martínez, O.; Garcia-Montero, C.; Alvarez-Mon, M.; Toledo-Lobo, M.d.V.; Ortega, M.A. Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes? Cancers 2023, 15, 4651. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK–Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Chen, Y.-C.; Kuo, C.-T.; Yu, H.W.; Chen, Y.-Q.; Chiou, A.; Kuo, J.-C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5, 18476. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.-Y.E.; Lim, S.-T.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Functional roles of SRC signaling in pancreatic cancer: Recent insights provide novel therapeutic opportunities. Oncogene 2023, 42, 1786–1801. [Google Scholar] [CrossRef] [PubMed]
- Peiró, G.; Ortiz-Martínez, F.; A Gallardo, A.; A Pérez-Balaguer, A.; Sánchez-Payá, J.; Ponce, J.J.; A Tibau, A.; López-Vilaro, L.; Escuin, D.; E Adrover, E.; et al. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br. J. Cancer 2014, 111, 689–695. [Google Scholar] [CrossRef]
- Beadnell, T.C.; Nassar, K.W.; Rose, M.M.; Clark, E.G.; Danysh, B.P.; Hofmann, M.-C.; Pozdeyev, N.; Schweppe, R.E. Src-mediated regulation of the PI3K pathway in advanced papillary and anaplastic thyroid cancer. Oncogenesis 2018, 7, 23. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Komlosh, P.G.; Chen, J.L.; Childs-Disney, J.; Disney, M.D.; Canaani, D. Broad-spectrum metastasis suppressing compounds and therapeutic uses thereof in human tumors. Sci. Rep. 2023, 13, 20420. [Google Scholar] [CrossRef]
- Tedeschi, A.; Nguyen, T.; Puttagunta, R.; Gaub, P.; Di Giovanni, S. A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 2009, 16, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Watabe, M.; Hirota, S.; Hosobe, S.; Miura, K.; Tegtmeyer, P.J.; Rinker-Shaeffer, C.W.; Watabe, K. The expression of the KAI1 gene, a tumor metastasis suppressor, is directly activated by p53. Proc. Natl. Acad. Sci. USA 1998, 95, 11307–11311. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.-H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.-P. MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Juan, L.-J.; Shia, W.-J.; Chen, M.-H.; Yang, W.-M.; Seto, E.; Lin, Y.-S.; Wu, C.-W. Histone Deacetylases Specifically Down-regulate p53-dependent Gene Activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar] [CrossRef]
- Wei, S.; Xiong, M.; Zhan, D.-Q.; Liang, B.-Y.; Wang, Y.-Y.; Gutmann, D.H.; Huang, Z.-Y.; Chen, X.-P. Ku80 functions as a tumor suppressor in hepatocellular carcinoma by inducing S-phase arrest through a p53-dependent pathway. Carcinogenesis 2012, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Kelman, Z. PCNA: Structure, functions and interactions. Oncogene 1997, 14, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Bar, J.; Lukaschuk, N.; Zalcenstein, A.; Wilder, S.; Seger, R.; Oren, M. The PI3K inhibitor LY294002 prevents p53 induction by DNA damage and attenuates chemotherapy-induced apoptosis. Cell Death Differ. 2005, 12, 1578–1587. [Google Scholar] [CrossRef]
- Cai, J.; Ye, Z.; Hu, Y.; Ye, L.; Gao, L.; Wang, Y.; Sun, Q.; Tong, S.; Zhang, S.; Wu, L.; et al. Fatostatin induces ferroptosis through inhibition of the AKT/mTORC1/GPX4 signaling pathway in glioblastoma. Cell Death Dis. 2023, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Gong, X.; Wang, Z.-Q.; Ding, Y.; Wang, C.; Luo, T.-F.; Piao, M.-H.; Meng, F.-K.; Chi, G.-F.; Luo, Y.-N.; et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol. Sin. 2017, 38, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Hung, M.-C. Nuclear EGFR signalling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Shi, Y.; Lv, Y.; Yuan, S.; Ramirez, C.F.A.; Lieftink, C.; Wang, L.; Wang, S.; Wang, C.; Dias, M.H.; et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021, 595, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Chen, Z.; Xu, Y. The double-edged functions of necroptosis. Cell Death Dis. 2023, 14, 163. [Google Scholar] [CrossRef]
- Meier, P.; Legrand, A.J.; Adam, D.; Silke, J. Immunogenic cell death in cancer: Targeting necroptosis to induce antitumour immunity. Nat. Rev. Cancer 2024. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Park, H.; Heisler, J.; Maculins, T.; Roose-Girma, M.; Xu, M.; Mckenzie, B.; Campagne, M.v.L.; Newton, K.; Murthy, A.; et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 2019, 8, e44452. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.-E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Welz, P.-S.; Wullaert, A.; Vlantis, K.; Kondylis, V.; Fernández-Majada, V.; Ermolaeva, M.; Kirsch, P.; Sterner-Kock, A.; van Loo, G.; Pasparakis, M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011, 477, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.; Prince, D.S.; Kench, C.; Liu, K. Alcohol and its associated liver carcinogenesis. J. Gastroenterol. Hepatol. 2023, 38, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Radike, M.J.; Stemmer, K.L.; Bingham, E. Effect of ethanol on vinyl chloride carcinogenesis. Environ. Health Perspect. 1981, 41, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Kushida, M.; Wanibuchi, H.; Morimura, K.; Kinoshita, A.; Kang, J.S.; Puatanachokchai, R.; Wei, M.; Funae, Y.; Fukushima, S. Dose-dependence of promotion of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline-induced rat hepatocarcinogenesis by ethanol: Evidence for a threshold. Cancer Sci. 2005, 96, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, B.; Ekström, T. The effects of long-term oral administration of ethanol on Sprague-Dawley rats—A condensed report. Toxicology 1995, 96, 133–145. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Niemann, P.; Schiffer, M.; Malan, D.; Grünberg, S.; Roell, W.; Geisen, C.; Fleischmann, B.K. Generation and Characterization of an Inducible Cx43 Overexpression System in Mouse Embryonic Stem Cells. Cells 2022, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-S.; Wu, L.-Q.; Yi, X.; Geng, C.; Li, Y.-J.; Yao, R.-Y. Connexin-43 can delay early recurrence and metastasis in patients with hepatitis B-related hepatocellular carcinoma and low serum alpha-fetoprotein after radical hepatectomy. BMC Cancer 2013, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Costa, C.J.S.; Pieters, A.; Rodrigues, B.d.S.; Van Campenhout, R.; Cooreman, A.; Tabernilla, A.; Cogliati, B.; Vinken, M. Expression and Functionality of Connexin-Based Channels in Human Liver Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 12187. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Pitchakarn, P.; Suzuki, S.; Chewonarin, T.; Tang, M.; Takahashi, S.; Naiki-Ito, A.; Sato, S.; Takahashi, S.; Asamoto, M.; et al. Silencing of connexin 43 suppresses invasion, migration and lung metastasis of rat hepatocellular carcinoma cells. Cancer Sci. 2012, 103, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Chiang, F.F.; Wang, H.M. Cetuximab plus chemotherapy as first-line treatment for metastatic colorectal cancer: Effect of KRAS mutation on treatment efficacy in Taiwanese patients. Neoplasma 2013, 60, 607–616. [Google Scholar] [CrossRef]
- Ding, Y.; Xiao, X.; Bai, L.; Yang, B.; Lin, G.; Zeng, L.; Xie, L.; Li, L.; Duan, X.; Shen, J.; et al. Enhanced radiosensitivity and chemoradiation efficacy in nasopharyngeal carcinoma via a dual-targeted SPION@polymer hybrid nanosensitizer. NPG Asia Mater. 2023, 15, 37. [Google Scholar] [CrossRef]
- Liu, W.; Cui, Y.; Wei, J.; Sun, J.; Zheng, L.; Xie, J. Gap junction-mediated cell-to-cell communication in oral development and oral diseases: A concise review of research progress. Int. J. Oral Sci. 2020, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; White, T.W.; Paul, D.L. Connections with Connexins: The Molecular Basis of Direct Intercellular Signaling. JBIC J. Biol. Inorg. Chem. 1996, 238, 1–27. [Google Scholar] [CrossRef]
- McCutcheon, S.; Stout, R.F.; Spray, D.C. The dynamic Nexus: Gap junctions control protein localization and mobility in distinct and surprising ways. Sci. Rep. 2020, 10, 17011. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Basilio, D.; Sáez, J.C.; Orellana, J.A.; Raine, C.S.; Bukauskas, F.; Bennett, M.V.L.; Berman, J.W. The Role of Gap Junction Channels During Physiologic and Pathologic Conditions of the Human Central Nervous System. J. Neuroimmune Pharmacol. 2012, 7, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Hirohashi, S. Cadherin intercellular adhesion molecule in hepatocellular carcinomas: Loss of E-cadherin expression in an undifferentiated carcinoma. Cancer Lett. 1991, 57, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.-H.; He, A.R.; Ryoo, B.-Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.S.; Di Cui, J.; Lee, D.; Yuen, V.W.-H.; Chiu, D.K.-C.; Goh, C.C.; Cheu, J.W.-S.; Tse, A.P.-W.; Bao, M.H.-R.; Wong, B.P.Y.; et al. Hypoxia-induced macropinocytosis represents a metabolic route for liver cancer. Nat. Commun. 2022, 13, 954. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Macias, R.I.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.; Glazer, P.M. Cell-interdependent cisplatin killing by Ku/DNA-dependent protein kinase signaling transduced through gap junctions. Proc. Natl. Acad. Sci. USA 2004, 101, 6134–6139. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Nguyen, T.A. Gap Junction Enhancer Increases Efficacy of Cisplatin to Attenuate Mammary Tumor Growth. PLoS ONE 2012, 7, e44963. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Connexin Studied and Reference | Material Studied | Expression—Regulation of Expression | Clinical Implication | Mechanisms Involved |
|---|---|---|---|---|
| Ma et al. (2002) [62] | Cell lines (HCC cell lines: HHCC, SMMC-7721 and normal liver cell line: QZG) [62] | 1. Low expression in HCC samples compared to normal liver samples [62] 2. Downregulation of expression during tumorigenesis because aberrant localization to the cytoplasm [62] | Not applicable | Not applicable |
| Sheen et al. (2004) [63] | Tissue samples (25 HCC samples and 15 normal controls) [63] | 1. Lower expression in HCC compared to normal cells [63] 2. Expression significantly correlated with cell differentiation [63] 3. No correlations between expression and gender, age, serum AFP level, chronic HBV or HCV carriage, tumor size, coexisting cirrhosis, encapsulation, vascular permeation, daughter nodules, tumor necrosis, or tumor hemorrhage [63] | Upregulated expression is associated with high recurrence end recurrence-related mortality [63] | Not applicable |
| Li et al. (2007) [64] | HuH7 cell lines with overexpressed Cx32 by doxycycline withdrawal (Tet-off HuH7 cells) and Li-7 HCC cell lines [64] | 1. Expression occurs mainly in the cytoplasm, but not in cell-to-cell contact areas, suggesting intracellular Cx32 sorting to the plasma membrane [64] 2. Overexpression of cytoplasmic Cx32 protein enhances proliferation, motility, and invasiveness in a gap junction independent manner [64] | Not applicable | Not applicable |
| Kawasaki et al. (2011) [65] | HuH7 HCC cell lines with overexpressed Cx32 by doxycycline withdrawal (Tet-off HuH7 cells) and HuH7 Tet-off mock cells [65] | 1. Higher Cx32 expression is associated with larger SP fractions [65] 2. Cytoplasmic accumulation of Cx32 expands CSC population development, elevating CSC renewal rate and enhancing spheres formation [65] | Not applicable | Not applicable |
| Yang et al. (2017) [66] | 1. Tissue samples (76 HCC samples and 20 normal controls) 2. Cell lines (normal hepatic cell line: LO2, HCC cell lines: HepG2, Huh7 and SMMC-77210) 3. Mouse models (Huh7-hCx32 cells or Huh7-vec cells inoculated) [66] | 1. Lower expression in HCC tissue compared to normal tissue [66] 2. Ectopic expression from the cell membrane (where is normally expressed) to the cytoplasm in HCC tissue [66] 3. Expression negatively correlated with histological grade and lymph node metastasis [66] 4. Expression not correlated with age, sex, tumor size, TNM stage, liver disease medical history, vascular embolus [66] | 1. Downregulation of expression is associated with a metastatic phenotype [66] 2. Downregulation associated with chemoresistance to oxaliplatin [66] 3. Targeting Cx32 proposed as a potential target to overcome oxaliplatin resistance [66] | 1. Downregulation of expression enhances cell migration and invasion [66] 2. Downregulation enhances EMT [66] 3. EMT regulated by Cx32 in HCC cells is mediated by Snail signaling pathway [66] |
| Yang et al. (2019) [59] | HepG2, Huh7, and SMMC-7721 cell lines (oxaliplatin-resistant cell lines development and parental HCC lines as controls) [59] | 1. High expression in the cell membrane in adjacent non-tumor tissues [59] 2. Low expression in the HCC cells, mainly expressed in the cytoplasm [59] 3. Low expression in oxaliplatin resistance cells [59] | 1. Downregulation associated with chemoresistance to oxaliplatin [59] 2. Targeting Cx32 proposed as a potential target to overcome oxaliplatin resistance [59] 4. Expression positively correlated to chemosensitivity to oxaliplatin [59] | 1. EMT phenotype and low Cx32 expression in oxaliplatin-resistant cells [59] 2. Downregulating Cx32 expression resulted in EMT induction [59] 3. Cx32 expression positively correlated with E-cadherin and negatively correlated with the expression of Vimentin and Snail [59] |
| Yu et al. (2017-1) [67] | 40 HCC tissue samples, HepG2 cell lines (oxaliplatin-resistant HepG2/DOX cell lines and parental HepG2 cell lines as controls) [67] | 1. Lower expression in HCC tissue compared to para-cancerous tissue [67] 2. Expression positively correlated with E-cadherin expression (in HCC tissues compared to para-cancerous tissue) and negatively correlated with Vimentin expression (higher in HCC tissue) [67] 3. Low expression in doxorubicin-resistant cells [67] 4. Expression may play a role in acquired drug resistance [67] | 1. Downregulation associated with chemoresistance to doxorubicin [67] 2. Targeting Cx32 proposed as a potential target to overcome doxorubicin resistance [67] | 1. Downregulating Cx32 expression resulted in EMT induction [67] 2. Cx32 expression was positively correlated with the expression of E-cadherin and negatively correlated with the expression of Vimentin [67] |
| Yu et al. (2017-2) [68] | 54 HCC tissues, HepG2 cell lines [68] | 1. Lower expression in HCC compared to para-cancerous normal liver tissues [68] 2. Expression positively correlated with differentiation degree [68] 3. Low expression in doxorubicin-resistant cells [68] | Downregulation associated with chemoresistance to doxorubicin Targeting Cx32 and Src/FAK signaling pathway is proposed as a potential target to overcome doxorubicin resistance [68] | Cx32 affects the chemoresistance to doxorubicin via the regulation of the activity of Src/FAK signaling pathway. Expression positively correlated to chemosensitivity to doxorubicin [68] |
| Zhao et al. (2015) [69] | 1.24 HCC tissue samples and 24 normal liver tissue samples [69] 2. Cell lines (HepG2, QGY-7701, SMMC-7721, MHCC97-H) [69] 3. Mouse models (inoculated with MHCC97H-shCtlr and MHCC97H-shCx32 cell lines) [69] | Lower expression in HCC tissue compared to normal tissue [69] | Downregulation associated with a poor prognosis [69] | 1. Cx32 expression suppressed invasion and migration via p53 pathway [69] 2. Cx32 upregulates CD82 expression via p53 [69] 3. Cx32 suppressed HCC progression in vivo [69] |
| Li et al. (2022) [70] | 1.85 HCC tissue samples Cell lines (HCCLM3 and HepG2) [70] 2. Mouse models (BALB/c) inoculated or not with HCCLM3 (HCCLM3 over expression—OE HCCLM3 empty vector—EV) [70] | 1. Expression downregulated in CSCs [70] 2. Expression regulated CSCs expansion [70] | Expression associated with a poor prognosis. Targeting Cx32 potentially inhibits the invasion and metastasis of liver cancer cells and reverses drug resistance [70] | Cx32 regulates the activity of the PI3K/Akt signaling pathway in HCC cells and CSCs expansion by the PI3K/Akt signaling pathway [70] |
| Xiang et al. (2019) [71] | 1. 96 HCC tissue samples 2. Cell lines (HepG2 and SMMC-7721) [71] | 1. Overexpression and internalization in HCC [71] 2. Expression positively correlated with Bcl-2 expression and negatively correlated with Bax and Bak expression [71] | 1. Expression associated with a poor prognosis [71] 2. Cx32 presents an intrinsic anti-apoptotic effect in HCC cells, in case of impaired gap junctions’ function [71] | Interaction between Cx32 and Src contributes to the EGFR activation which mediates the Cx32 anti-apoptotic effect in HCC cells [71] |
| Xiang et al. (2021) [72] | 1. HCC tissue samples [72] 2. PLC/PRF/5 and SMMC-7721 cell lines [72] 3. Mouse models (BALB/c-nu mice) inoculated with PLC-Vector cells and PLC-shCx32 cells (PLC-Vector+Vehicle, PLC- shCx32+Vehicle, PLC-Vector+SHN, and PLC-shCx32+SHN) [72] | 1. Expression positively correlated with expression levels of necroptosis biomarkers (RIP1, p-RIP1, and p-MLKL) [72] 2. Downregulated expression suppresses SHN-induced necroptosis in PLC/PRF/5 cells [72] 3. Upregulated expression enhances SHN-induced necroptosis by upregulation of RIP1, RIP3, and MLKL in HCC cells [72] | 1. Cx32 may be involved in a therapeutic strategy consisting of necroptosis inducers, which may be effective in HCC patients with high Cx32 expression levels [72] 2. The overexpression of Cx32 could be a potential therapeutic biomarker in HCC [72] | 1. Cx32 enhances the c-FLIPs and downregulates FADD, resulting in caspase 8 inactivation and protection from RIP1 and RIP3 caspase 8-mediated cleavage [72] 2. Cx32 interacts with Src and contributes to the Src-mediated phosphorylation of caspase 8, resulting in suppression of caspase 8 and activation of necroptosis [72] 3. Cx32 knockdown suppresses necroptosis in vivo [72] |
| Kato et al. (2016) [73] | Mouse models: -Cx32 dominant negative transgenic (Tg) -wild-type (Wt), which were given 1% or 5% ethanol or water ad libitum for 16 weeks after an intraperitoneal injection of 200 mg/kg diethyl nitrosamine [73] | Downregulation of expression positively correlated with Dusp1 and Dusp4 downregulation of expression in a protein level and with Dusp1 downregulation of expression in a mRNA level, in Tg mouse models [73] | Dysregulated expression may promote ethanol-related hepato-carcinogenesis [73] | Cx32 dysfunction compared with exposure to ethanol decreases Dusp1 expression leading to Erk activation in GST-P positive foci, enhancing tumorigenic activity [73] |
| Sagawa et al. (2015) [74] | Mouse models: -Cx32 dominant negative transgenic (Cx32ΔTg) -wild-type (Wt), which were given diethylnitrosamine and fed methionine–choline-deficient diet (MCDD) or MCDD with luteolin for 12 weeks [74] | Expression negatively correlated with NASH development [74] | 1. Dysregulated expression may promote steatohepatitis and fibrosis [74] 2. Reduced expression of Cx32 associated with NASH is prevented by luteolin [74] | Not applicable |
| Connexin Studied and Reference | Material Studied | Expression | Clinical Implication | Mechanisms Involved |
|---|---|---|---|---|
| Ma et al. (2002) [62] | Cell lines (HCC cell lines: HHCC, SMMC-7721 and normal liver cell line: QZG) [62] | 1. Low expression in HCC samples compares to normal liver samples, except expression in SMMC-7721 cells [62] 2. mRNA expression not significantly different between HCC and normal liver samples [62] | Not applicable | Not applicable |
| Sheen et al. (2004) [63] | Tissue samples (25 HCC samples and 15 normal controls) [63] | Higher expression in HCC tissue samples [63] | Expression not correlated with recurrence and mortality [63] | Not applicable |
| Wang et al. (2013) [149] | 38 HBV-HCC tissue samples [149] | 1. Expression positively correlated with histological differentiation, multiple foci, and vascular tumor thrombosis [149] 2. Expression negatively correlated with MVD-CD105 and VEGF expression levels, and with distant metastases [149] | 1. Cx43 downregulated expression associated with poor prognosis in HBV-HCC patients with a low AFP level [149] 2. Survival rates are positively correlated with Cx43 expression [149] | Not applicable |
| Ogawa et al. (2012) [151] | 1. HSU-C1, C5F, -C6, -N1 and -L2 cell lines [151] 2. Mouse models (inoculated with C5F, C6, N1 and L2 cells) [151] | 1. Higher expression in high metastatic cell lines (N1 and L1) compared with low metastatic cell lines (C1, C5F, C6) [151] 2. Higher expression endothelial cells–tumor cells contact areas [151] | 1. Silencing with siRNA resulted in decrease of lung metastasis in mouse models [151] 2. Silencing with siRNA suppressed invasion and migration of L2 cell line (accompanied by MMP-9 decrease) [151] | 1. MMP-9 association with HCC metastatic capacity explains the decreased metastatic capacity as a result of MMP-9 decrease via Cx-43 silencing [151] 2. Higher expression of Cx-43 in highly metastatic HCC cells results in GJ formation between tumor cells and endothelial cells [151] |
| Wang et al. (2019) [152] | 80 HCC tissue samples Cell lines (SMMC-7221) [152] | Expression negatively associated with RALA and SRC genes expression [152] | Targeting the downstream mechanisms regulated by Cx43 (RALA, SRC: target genes) is a potential therapeutic strategy for advanced HCC [152] | RALA and SRC are significantly upregulated in CX43-silenced HCC cells and significantly associated with HCC survival [152] |
| Connexin Studied and Reference | Material Studied | Expression—Regulation of Expression | Mechanisms Involved | Clinical Implication |
|---|---|---|---|---|
| Sheen et al. (2004) [63] | HCC tissue samples (25) compared to normal controls (15) [63] | 1. Lower expression in HCC compared to normal cells.Expression significantly correlated with cell differentiation [63] 2. No correlations between expression and gender, age, serum AFP level, chronic HBV or HCV carriage, tumor size, coexisting cirrhosis, encapsulation, vascular permeation, daughter nodules, tumor necrosis, or tumor hemorrhage [63] | Not applicable | Not applicable |
| Li et al. (2021) [22] | HCC tissue and cell lines (pG2 with low Cx26 expression and SK-hep-1 with high Cx26 expression [22] | Same expression in irradiated and control cells Expression positively associated with survival [22] | Overexpression positively correlated with overactivation of MAPK and NF-κB signaling pathways [22] | Expression positively correlated with radiosensitivity [22] |
| Yang et al. (2016) [23] | Cell lines (normal liver cell line: LO2 and HCC cell lines: SMMC-7721) [23] | Endothelial growth factor and increased adherent proteins affect chemosensitivity [23] | Cx26 inhibition decreases oxaliplatin cytotoxicity [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakos, S.P.; Chatzikalil, E.; Arvanitakis, K.; Vakadaris, G.; Stergiou, I.E.; Koutsompina, M.-L.; Argyrou, A.; Lekakis, V.; Konstantinidis, I.; Germanidis, G.; et al. Understanding the Role of Connexins in Hepatocellular Carcinoma: Molecular and Prognostic Implications. Cancers 2024, 16, 1533. https://doi.org/10.3390/cancers16081533
Papadakos SP, Chatzikalil E, Arvanitakis K, Vakadaris G, Stergiou IE, Koutsompina M-L, Argyrou A, Lekakis V, Konstantinidis I, Germanidis G, et al. Understanding the Role of Connexins in Hepatocellular Carcinoma: Molecular and Prognostic Implications. Cancers. 2024; 16(8):1533. https://doi.org/10.3390/cancers16081533
Chicago/Turabian StylePapadakos, Stavros P., Elena Chatzikalil, Konstantinos Arvanitakis, Georgios Vakadaris, Ioanna E. Stergiou, Maria-Loukia Koutsompina, Alexandra Argyrou, Vasileios Lekakis, Ippokratis Konstantinidis, Georgios Germanidis, and et al. 2024. "Understanding the Role of Connexins in Hepatocellular Carcinoma: Molecular and Prognostic Implications" Cancers 16, no. 8: 1533. https://doi.org/10.3390/cancers16081533
APA StylePapadakos, S. P., Chatzikalil, E., Arvanitakis, K., Vakadaris, G., Stergiou, I. E., Koutsompina, M. -L., Argyrou, A., Lekakis, V., Konstantinidis, I., Germanidis, G., & Theocharis, S. (2024). Understanding the Role of Connexins in Hepatocellular Carcinoma: Molecular and Prognostic Implications. Cancers, 16(8), 1533. https://doi.org/10.3390/cancers16081533