Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells

 and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Lines and Transformants
2.2. RNA Expression Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5' to 3') | Reverse Primer (5' to 3') |
|---|---|---|
| Primers for qRT-PCR | ||
| Actin | ACCGCGAGAAGATGACCCAG | AGGTCCAGACGCAGGATGG |
| EpCAM | CGCAGCTCAGGAAGAATGTG | TGAAGTACACTGGCATTGACGA |
| ESRP1 | TCCTGCTGTTCTGGAAAGTCG | TCCGGTCTAACTAGCACTTCGTG |
| CDH1 * | CGGGAATGCAGTTGAGGATC | AGGATGGTGTAAGCGATGGC |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
| NRP2 | GGATGGCATTCCACATGTTG | ACCAGGTAGTAACGCGCAGAG |
| RAB25 | AATGTTCGCTGAAAACAATGGAC | CTCAAAGGCTAGCTCAACATTGG |
| SEMA3F | AGCAGACCCAGGACGTGAG | AAGACCATGCGAATATCAGCC |
| ST14 | GGGACACACCCAGTATGGAGG | GAGGTTCTCGCAGGTGGTCTG |
| ZEB1 | AGCAGTGAAAGAGAAGGGAATGC | GGTCCTCTTCAGGTGCCTCAG |
| Primers for ChIP qPCR | ||
| Alu | GCCTGT AATCCCAGCACTTT | AAGCGATTCTCCTGCCTCAGC |
| EpCAM | TAGCCTCCACGTTCCTCTATCC | TGCTGAGACTTCCTTTTAACCG |
| ESRP1 | TCAGTCCTCCGCAACTTAGCT | TCCGAGACCCCACCTCGT |
| CDH1 | GGCCGGCAGGTGAACCCTCA | GGGCTGGAGTCTGAACTGA |
| NRP2 | TTAACCCACCCTGGAGTCTCC | GCAATAGCTGCTAATTTGAGCG |
| RAB25 | CACCCAACCTGTCGAACCT | GAGAGGACGGAAGCTGAGAAC |
| SEMA3F-pr | GGCGTATGGATGTGTGGATGA | TATGAGAGCACCCACCCAGAAC |
| SEMA3F-neg | CCCTACAGTTCCAGCAGCCC | CCACCAACCCAGACCCTGAT |
| ST14-pr | CAAAGTGAGCAAGGTGAAGGG | TTTATCCACCTCCTTGATGCC |
| ST14-neg | ATCTCCCACCCCTTCTTCAATG | GCTGTACTCTGCCGGTTTCTC |
2.3. MiRNA Analysis
2.4. Chromatin Immunoprecipitation (ChIP) Assay
| Primary antibodies | Reference | Lot | ChIP | IF | WB | TMA |
|---|---|---|---|---|---|---|
| Rabbit control serum | Sigma I5006 | 10 µg | ||||
| Rabbit anti-ZEB1 | Santa Cruz Biotechnology, H102 | D2010 | 10 µg | 1:50 | 1:50 | |
| Mouse monoclonal anti-ZEB1 | R&D MAB 6708 | CEZB0111021 | 1:50 | |||
| Rabbit anti-acetyl histone H3K9 | Active Motive 39137 | 01008001 | 10 µL | |||
| Rabbit anti-acetyl histone H3K27 | Active Motive 39133 | 3161003 | 10 µg | 1:500 | 1:20,000 | 1:50 |
| Rabbit anti-acetyl histone H3 | Millipore 06-599 | DAM1823380 | 10 µg | 1:500 | 1:20,000 | 1:50 |
| Rabbit anti-acetyl histone H4 | Millipore 06-866 | JBC1873473 | 10 µg | 1:20,000 | ||
| Rabbit monoclonal anti-E-cadherin | Cell Signaling 24E10 | 2 | 1:100 | 1:1,000 | ||
| Mouse anti-E-cadherin | B&D 610181 | 85521 | 1:100 | |||
| Anti-Rab25 | Sigma R8532 | 1:1,000 | ||||
| Anti-actin | Sigma | 1:50,000 |
2.5. Protein Detection by Immunoblotting
2.6. Immunofluorescence
2.7. Lung Cancer Tissue Microarray (TMA), Immuno-Histochemistry and Analysis
2.8. Statistical Analysis
3. Results
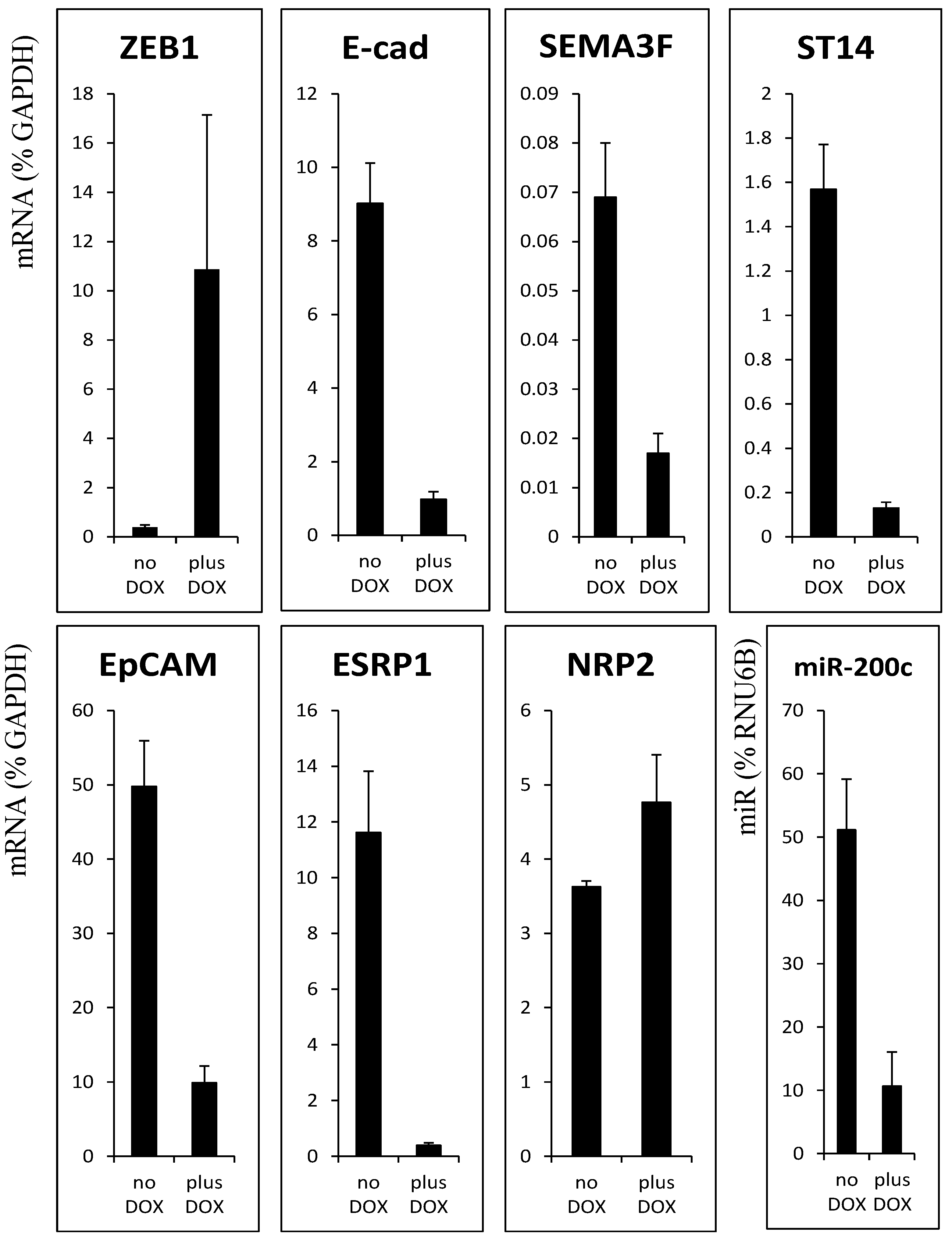
3.1. ZEB1 Binding to New Target Gene Promoter Regions



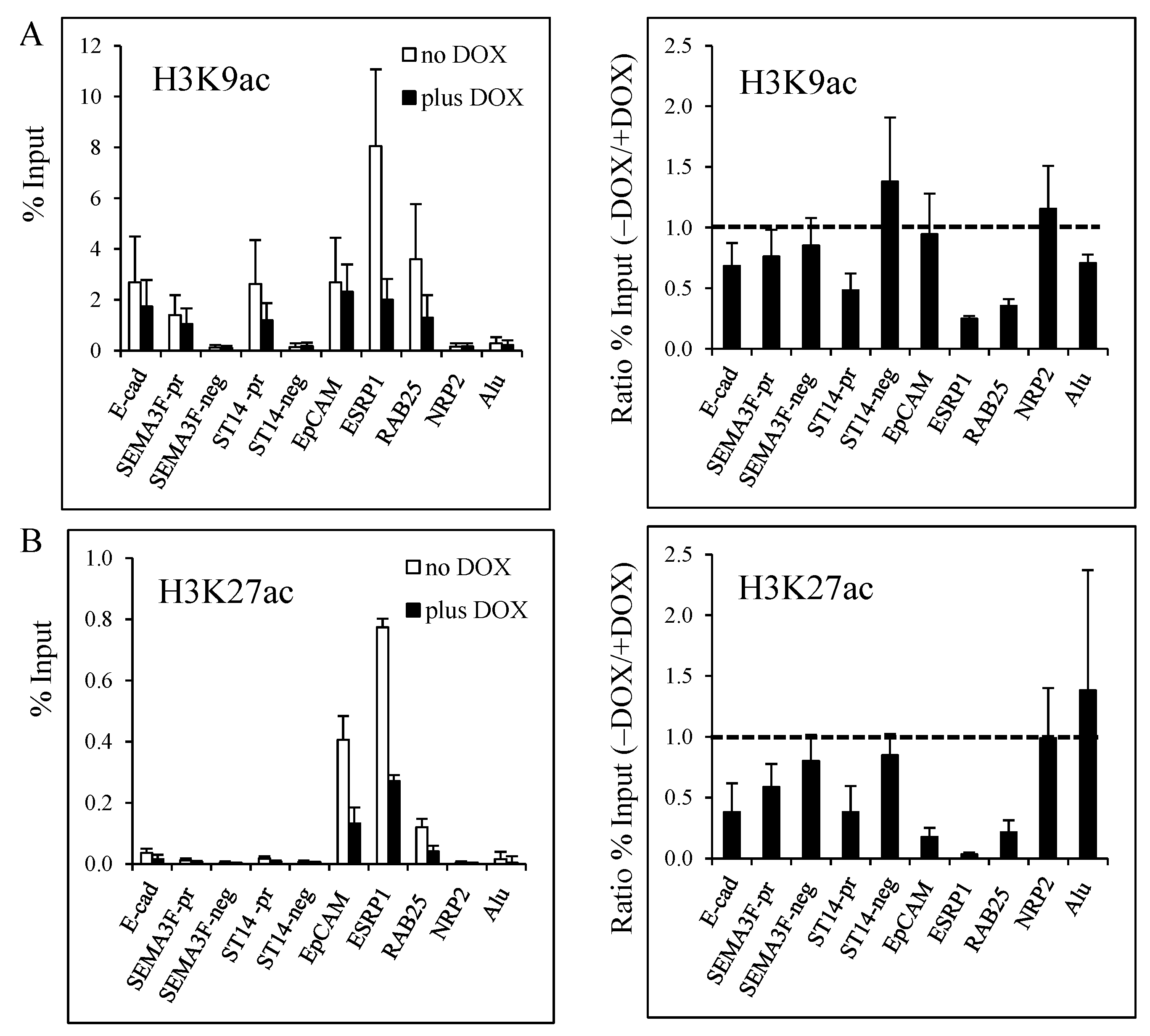
3.2. Decrease of Histone Acetylation by ZEB1 on Target Genes


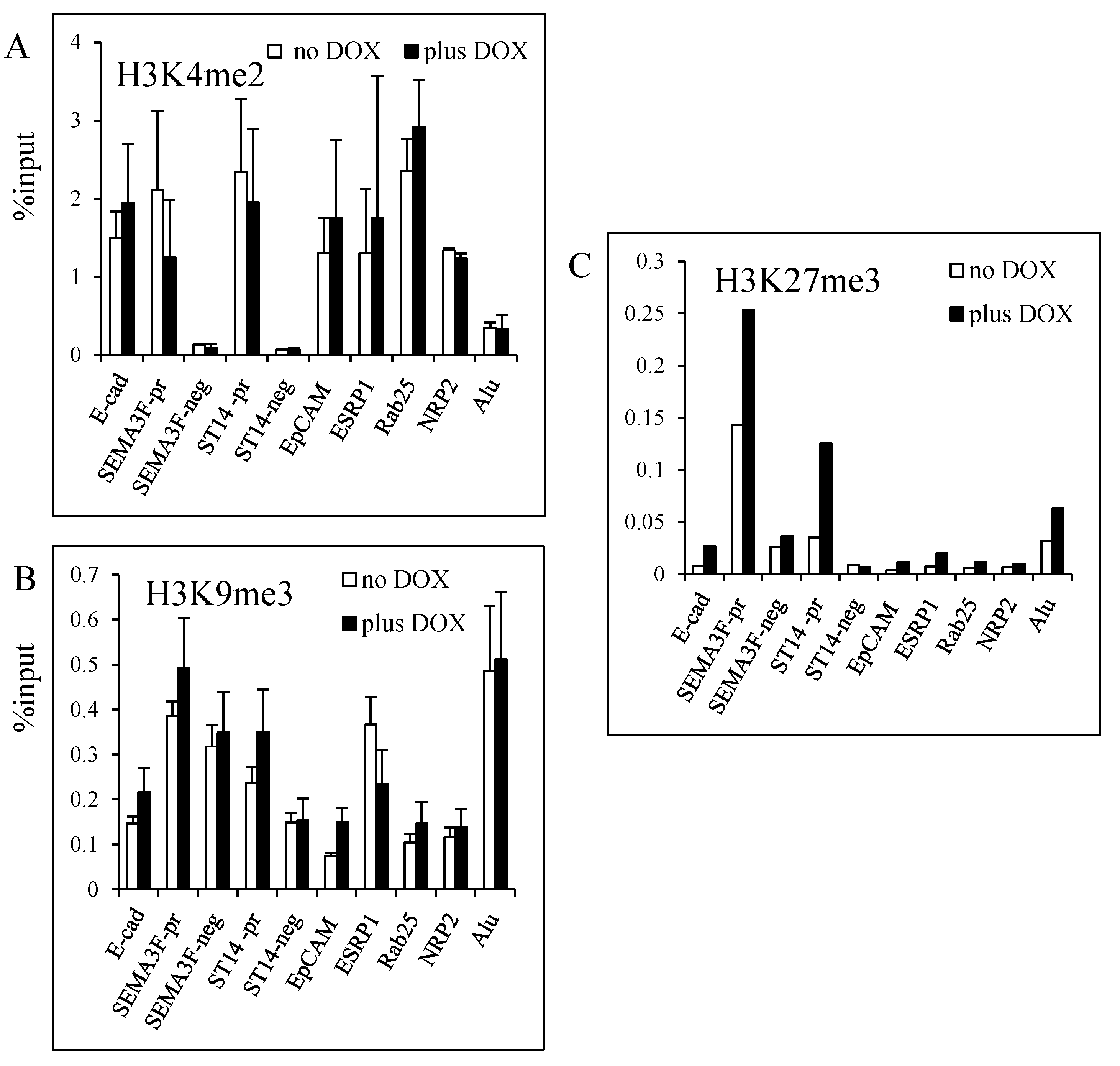
3.3. Decrease of Histone H3K27 Acetylation by ZEB1
3.4. Modulation of Histone H3K27 Methylation by ZEB1


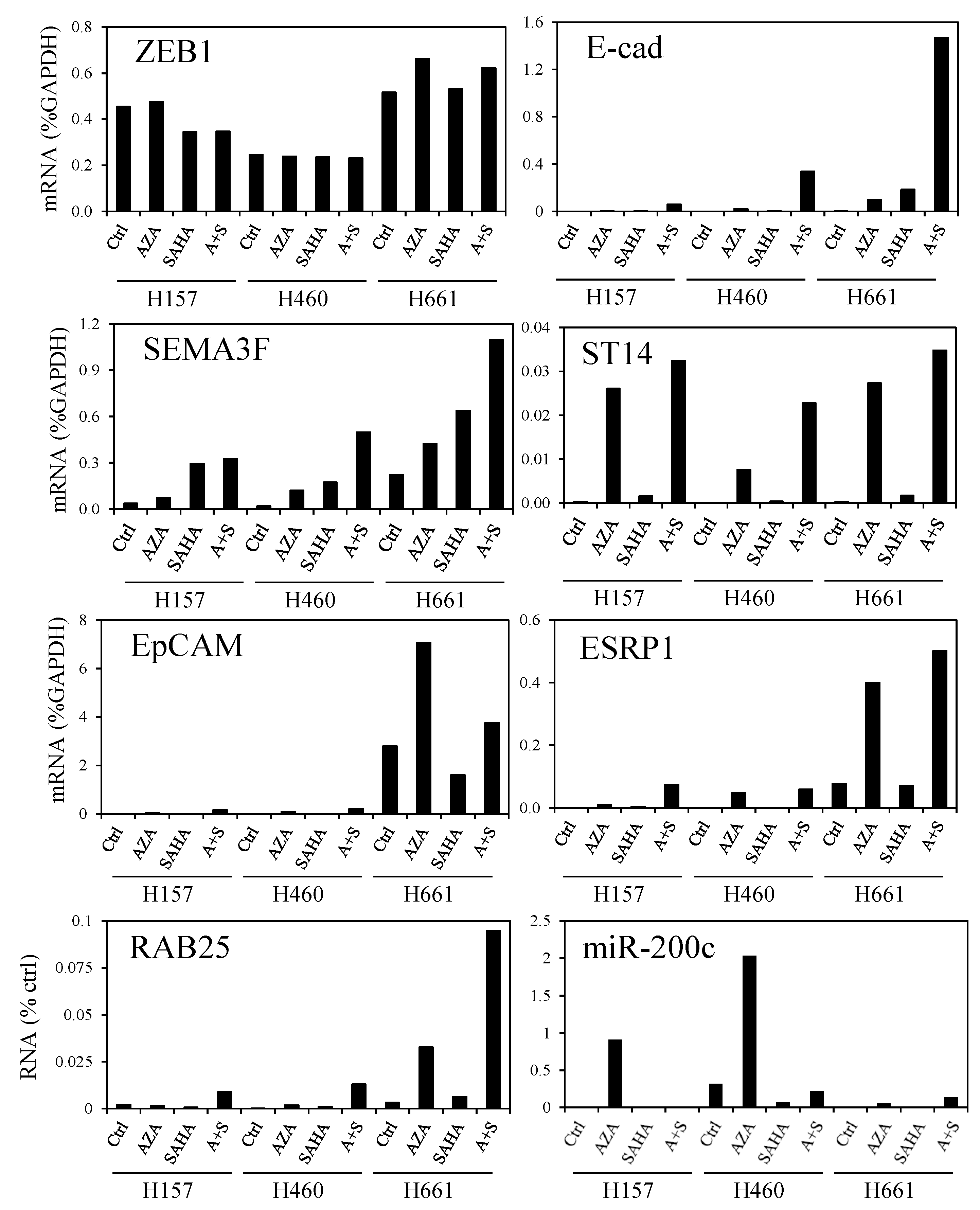
3.5. DNA Demethylation Agent and Histone Deacetylase Inhibitor Increase ZEB1 Target Gene Expression

3.6. Histone H3K27 Acetylation in Lung Cancers

4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
References
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Sabbah, M.; Emami, S.; Redeuilh, G.; Julien, S.; Prevost, G.; Zimber, A.; Ouelaa, R.; Bracke, M.; de Wever, O.; Gespach, C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist. Updat. 2008, 11, 123–151. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; de Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell. Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef]
- Ohira, T.; Gemmill, R.M.; Ferguson, K.; Kusy, S.; Roche, J.; Brambilla, E.; Zeng, C.; Baron, A.; Bemis, L.; Erickson, P.; et al. WNT7a induces E-cadherin in lung cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 10429–10434. [Google Scholar] [CrossRef]
- Takeyama, Y.; Sato, M.; Horio, M.; Hase, T.; Yoshida, K.; Yokoyama, T.; Nakashima, H.; Hashimoto, N.; Sekido, Y.; Gazdar, A.F.; et al. Knockdown of ZEB1, a master epithelial-to-mesenchymal transition (EMT) gene, suppresses anchorage-independent cell growth of lung cancer cells. Cancer Lett. 2010, 296, 216–224. [Google Scholar] [CrossRef]
- Clarhaut, J.; Gemmill, R.M.; Potiron, V.A.; Ait-Si-Ali, S.; Imbert, J.; Drabkin, H.A.; Roche, J. ZEB-1, a repressor of the semaphorin 3F tumor suppressor gene in lung cancer cells. Neoplasia 2009, 11, 157–166. [Google Scholar]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2010, 300, 66–78. [Google Scholar]
- Tang, B.L.; Ng, E.L. Rabs and cancer cell motility. Cell Motil. Cytoskeleton 2009, 66, 365–370. [Google Scholar] [CrossRef]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 2012, 22, 131–145. [Google Scholar] [CrossRef]
- Goldenring, J.R.; Nam, K.T. Rab25 as a tumour suppressor in colon carcinogenesis. Br. J. Cancer 2011, 104, 33–36. [Google Scholar] [CrossRef]
- Agarwal, R.; Jurisica, I.; Mills, G.B.; Cheng, K.W. The emerging role of the RAB25 small GTPase in cancer. Traffic 2009, 10, 1561–1568. [Google Scholar] [CrossRef]
- Barrios-Rodiles, M.; Brown, K.R.; Ozdamar, B.; Bose, R.; Liu, Z.; Donovan, R.S.; Shinjo, F.; Liu, Y.; Dembowy, J.; Taylor, I.W.; et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005, 307, 1621–1625. [Google Scholar] [CrossRef]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Siles, L.; de Barrios, O.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Postigo, A. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am. J. Cancer Res. 2011, 1, 897–912. [Google Scholar]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef]
- Chinnadurai, G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 2002, 9, 213–224. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.; Sui, G.; Affar el, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Bunn, P.A., Jr. A new generation of EGFR tyrosine-kinase inhibitors in NSCLC. Lancet Oncol. 2012, 13, 442–443. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Gridelli, C.; de Marinis, F.; di Maio, M.; Cortinovis, D.; Cappuzzo, F.; Mok, T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating Epidermal Growth Factor Receptor mutation: Implications for clinical practice and open issues. Lung Cancer 2011, 72, 3–8. [Google Scholar] [CrossRef]
- Coldren, C.D.; Helfrich, B.A.; Witta, S.E.; Sugita, M.; Lapadat, R.; Zeng, C.; Baron, A.; Franklin, W.A.; Hirsch, F.R.; Geraci, M.W.; et al. Baseline gene expression predicts sensitivity to gefitinib in non-small cell lung cancer cell lines. Mol. Cancer Res. 2006, 4, 521–528. [Google Scholar] [CrossRef]
- Rizzolio, S.; Tamagnone, L. Multifaceted role of neuropilins in cancer. Curr. Med. Chem. 2011, 18, 3563–3575. [Google Scholar] [CrossRef]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef]
- Sato, M.; Shames, D.S.; Hasegawa, Y. Emerging evidence of Epithelial-to-Mesenchymal Transition in lung carcinogenesis. Respirology 2012, 17, 1048–1059. [Google Scholar] [CrossRef]
- Hou, J.M.; Krebs, M.; Ward, T.; Sloane, R.; Priest, L.; Hughes, A.; Clack, G.; Ranson, M.; Blackhall, F.; Dive, C. Circulating tumor cells as a window on metastasis biology in lung cancer. Am. J. Pathol. 2011, 178, 989–996. [Google Scholar] [CrossRef]
- Caswell, P.T.; Spence, H.J.; Parsons, M.; White, D.P.; Clark, K.; Cheng, K.W.; Mills, G.B.; Humphries, M.J.; Messent, A.J.; Anderson, K.I.; et al. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell 2007, 13, 496–510. [Google Scholar] [CrossRef]
- Cheng, K.W.; Lahad, J.P.; Kuo, W.L.; Lapuk, A.; Yamada, K.; Auersperg, N.; Liu, J.; Smith-McCune, K.; Lu, K.H.; Fishman, D.; et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 2004, 10, 1251–1256. [Google Scholar]
- Cheng, J.M.; Volk, L.; Janaki, D.K.; Vyakaranam, S.; Ran, S.; Rao, K.A. Tumor suppressor function of Rab25 in triple-negative breast cancer. Int. J. Cancer 2010, 126, 2799–2812. [Google Scholar]
- Schneider, G.; Kramer, O.H.; Saur, D. A ZEB1-HDAC pathway enters the epithelial to mesenchymal transition world in pancreatic cancer. Gut 2012, 61, 329–330. [Google Scholar] [CrossRef]
- Saunders, L.R.; Verdin, E. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar] [CrossRef]
- Kakihana, M.; Ohira, T.; Chan, D.; Webster, R.B.; Kato, H.; Drabkin, H.A.; Gemmill, R.M. Induction of E-cadherin in lung cancer and interaction with growth suppression by histone deacetylase inhibition. J. Thorac. Oncol. 2009, 4, 1455–1465. [Google Scholar] [CrossRef]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef]
- Lopez-Serra, P.; Esteller, M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene 2011, 29, 1609–1622. [Google Scholar]
- Schliekelman, M.J.; Gibbons, D.L.; Faca, V.M.; Creighton, C.J.; Rizvi, Z.H.; Zhang, Q.; Wong, C.H.; Wang, H.; Ungewiss, C.; Ahn, Y.H.; et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011, 71, 7670–7682. [Google Scholar] [CrossRef]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Mink, S.R.; Vashistha, S.; Zhang, W.; Hodge, A.; Agus, D.B.; Jain, A. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol. Cancer Res. 2010, 8, 809–820. [Google Scholar] [CrossRef]
- Van den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Khochbin, S.; Gazzeri, S. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 7237–7245. [Google Scholar] [CrossRef]
- Van den Broeck, A.; Ozenne, P.; Eymin, B.; Gazzeri, S. Lung cancer: A modified epigenome. Cell Adh. Migr. 2010, 4, 107–113. [Google Scholar] [CrossRef]
- Barlesi, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef]
- Tseng, R.C.; Lee, C.C.; Hsu, H.S.; Tzao, C.; Wang, Y.C. Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients. Neoplasia 2009, 11, 763–770. [Google Scholar]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roche, J.; Nasarre, P.; Gemmill, R.; Baldys, A.; Pontis, J.; Korch, C.; Guilhot, J.; Ait-Si-Ali, S.; Drabkin, H. Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers 2013, 5, 334-356. https://doi.org/10.3390/cancers5020334
Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, Guilhot J, Ait-Si-Ali S, Drabkin H. Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers. 2013; 5(2):334-356. https://doi.org/10.3390/cancers5020334
Chicago/Turabian StyleRoche, Joëlle, Patrick Nasarre, Robert Gemmill, Aleksander Baldys, Julien Pontis, Christopher Korch, Joëlle Guilhot, Slimane Ait-Si-Ali, and Harry Drabkin. 2013. "Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells" Cancers 5, no. 2: 334-356. https://doi.org/10.3390/cancers5020334
APA StyleRoche, J., Nasarre, P., Gemmill, R., Baldys, A., Pontis, J., Korch, C., Guilhot, J., Ait-Si-Ali, S., & Drabkin, H. (2013). Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers, 5(2), 334-356. https://doi.org/10.3390/cancers5020334