Clinicopathologic Characteristics and Outcomes of Histiocytic and Dendritic Cell Neoplasms: The Moffitt Cancer Center Experience Over the Last Twenty Five Years

Abstract
:1. Introduction and Methods
1.1. Introduction
1.2. Methods
2. Results
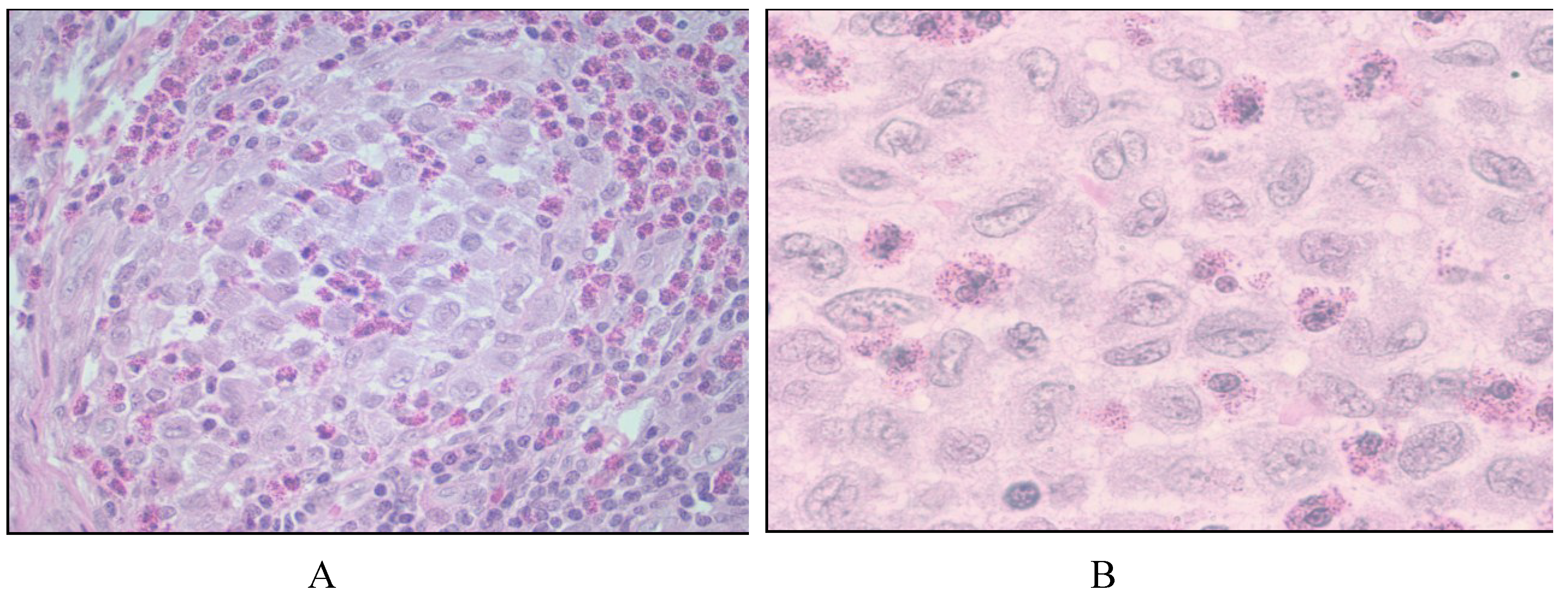
2.1. Langerhans Cell Histiocytosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Year of Diagnosis | Age at Diagnosis (Year)/Sex | Disease Subtype | Initial Site of Involvement | Initial Staging CT Scans/Bone Marrow (Yes/No) | Recurrence (Yes/No) | Immunohistochemistry | Vital Status | Overall Survival (months) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2010 | 18/F | LCH | Lungs/Mandible | No/No | No | NR | Alive | 41 |
| 2 | 2011 | 26/M | LCH | Lungs/Skin (nose) | Yes/No | No | Cd1a, CD68, S100 | Alive | 6 |
| 3 | 2011 | 16/F | LCH | Right Femur | No/No | No | CD1a, S100, CD68 | Alive | 20 |
| 4 | 2008 | 26/F | LCH | Lung/Bone/C2 spine | Yes/No | NR | CD1a, vimentin, S100, CD68, CD31 (focal), LCA (focal) (CD20-, CD30-, Cd99-, CD117-, melan-A-) | Alive | 30 |
| 5 | 2012 | 53/F | LCH | Right temporal bone | Yes/No | No | CD1a, S100 | Alive | 14 |
| 6 | 2010 | 51/F | LCH | Skin (nose) | Yes/Yes | NR | CD1a, (HMB-45-, CD117-, CD45-, CD3-, CD20-) | Alive | 1 |
| 7 | 2007 | 22/M | LCH | Ischial tuberosity | Yes/Yes | Yes | CD1a, CD68, S100 (CD3-, CD20-, CD5-, Cd15-, CD30-, PAX5-, MUM1-) | Alive | 77 |
| 8 | 2011 | 59/F | LCH | Skin (buccal) | Yes/No | No | CD1a, S100, CD43 (CD20-, CD3-,Cd30-, MPO-, HMB-45-, CK 5/6-, P63-) | Alive | 20 |
| 9 | 2008 | 60/F | LCH | Cecum/Lymph Nodes | Yes/Yes | No | CD1a, CD68, S100 (melan-A−, CD117−, HMB45−) | Alive | 64 |
| 10 | 2005 | 17/M | LCH | Lung/Bone/Pituitary/Hypothalamus | Yes/No | No | CD1a, S100 | Dead | 75 |
| 11 | 2007 | 42/F | LCH | Lung/T3 | Yes/Yes | Yes | CD1a, S100, CD68 | Alive | 61 |
| 12 | 2009 | 38/F | LCH | Parotid Gland | Yes/Yes | No | CD1a, CD68, S100 | Dead | 12 |
| 13 | 2008 | 61/M | LCH | Lymph Nodes/Inguinal lymph node | Yes/Yes | No | CD1a, CD4, S100, Langerin, CD68 weak positive, CD30 weak positive, Ki-67 30%−40%, (Alk-1−, panmelanoma−, oscar−, CD20−, CD21−, CD23−) | Alive | 3 |
| 14 | 2005 | 25/F | LCH | Skin (scalp) | Yes/Yes | No | CD1a, CD68, S100 | Alive | 102 |
| 15 | 1996 | 25/F | LCH | Right frontal lobe | Yes/Yes | No | NR | Alive | 191 |
| 16 | 2011 | 33/F | LCH | Skin (shoulder, face) | Yes/No | No | CD1a, S100, (-CD68, -melanA) | Alive | 26 |
| 17 | 2007 | 43/M | LCH | Seventh rib | No/No | No | CD1a, CD68, S100 (Pankeratin−) | Alive | 76 |
| 18 | 2009 | 71/F | LCH | Lung/bone marrow | Yes/Yes | No | CD1a, S100, (CD68−, lysozyme−, MPO−) | Dead | 9 |
| 19 | 2012 | 55/M | LCH | Skin (arm, back) | Yes/Yes | No | CD1a, CD68, S100, (CD 34−) | Alive | 5 |
| 20 | 1985 | 3/M | LCH | Bone marrow/lymph node | NR/Yes | Yes | NR | Alive | 347 |
| 21 | 2005 | 69/F | HS | Intestine | Yes/No | Yes | CD4, CD45RO, CD45, CD68, Vimentin, CD163 (S100−) | Dead | 52 |
| 22 | 2013 | 52/M | HS | Brain | Yes/No | No | NR | Dead | 5 |
| 23 | 1995 | 56/F | HS | Bone marrow | NR/Yes | No | NR | Alive | 221 |
| 24 | 2003 | 51/F | HS | Neck lymph node | Yes/No | No | CD68, Vimentin, Lysozyme, S100 (LCA−, CD30−, CK20−, CAM5.2−, TTF-1−, HMB-45−, CD34−, SMA−) | Alive | 101 |
| 25 | 1992 | 51/M | HS | Skin (thumb) | NR/NR | No | S100(−), LCA(−), Vimentin | ||
| 26 | 1994 | 68/M | HS | Bone marrow | NR/Yes | NR | NR | Dead | 0 |
| 27 | 2005 | 42/M | FRCT | Inguinal lymph node/nodes | Yes/Yes | No | CD14, S100, CD45, SMA (CD21−, CD35−, CD20−, CD10−, CD3−, AE/CAM5.2−)EMA | Alive | 51 |
| 28 | 2008 | 77/M | FDCS | Abdominal mass | Yes/NR | Yes | CD 21, CD23, CD68, D-240 (CD117−, desmin−, S-100−) | Dead | 22 |
| 29 | 2014 | 46/F | FDCS | Liver and lymph nodes | Yes/No | No | CD21, CD23, CD68 weak, S100+, CD138−, AE1/AE/CAM5.2− | Alive | 2 |
| 30 | 2008 | 90/F | IDCS | Soft tissue (anterior chest) | NR/NR | NR | CD68, S100, Vimentin (CD15−, CD30−, CD20−, CD3−, MART-1−, CD34−) | NR | NR |
| 31 | 2008 | 50/M | IDCS | Cervical lymph node | Yes/Yes | No | S100, Vimentin, CD68 (CD35−, CD21−, CD3−, CD20−, CD34−, SMA−, AE1/AE3−, CAM5d3−, Melan-A−, Ki low). | Alive | 58 |
| 32 | 2008 | 45/F | INDCS | Skin (supraclavicular) | Yes/Yes | No | CD1a, CD68, S100 (CD30−) | Alive | 13 |
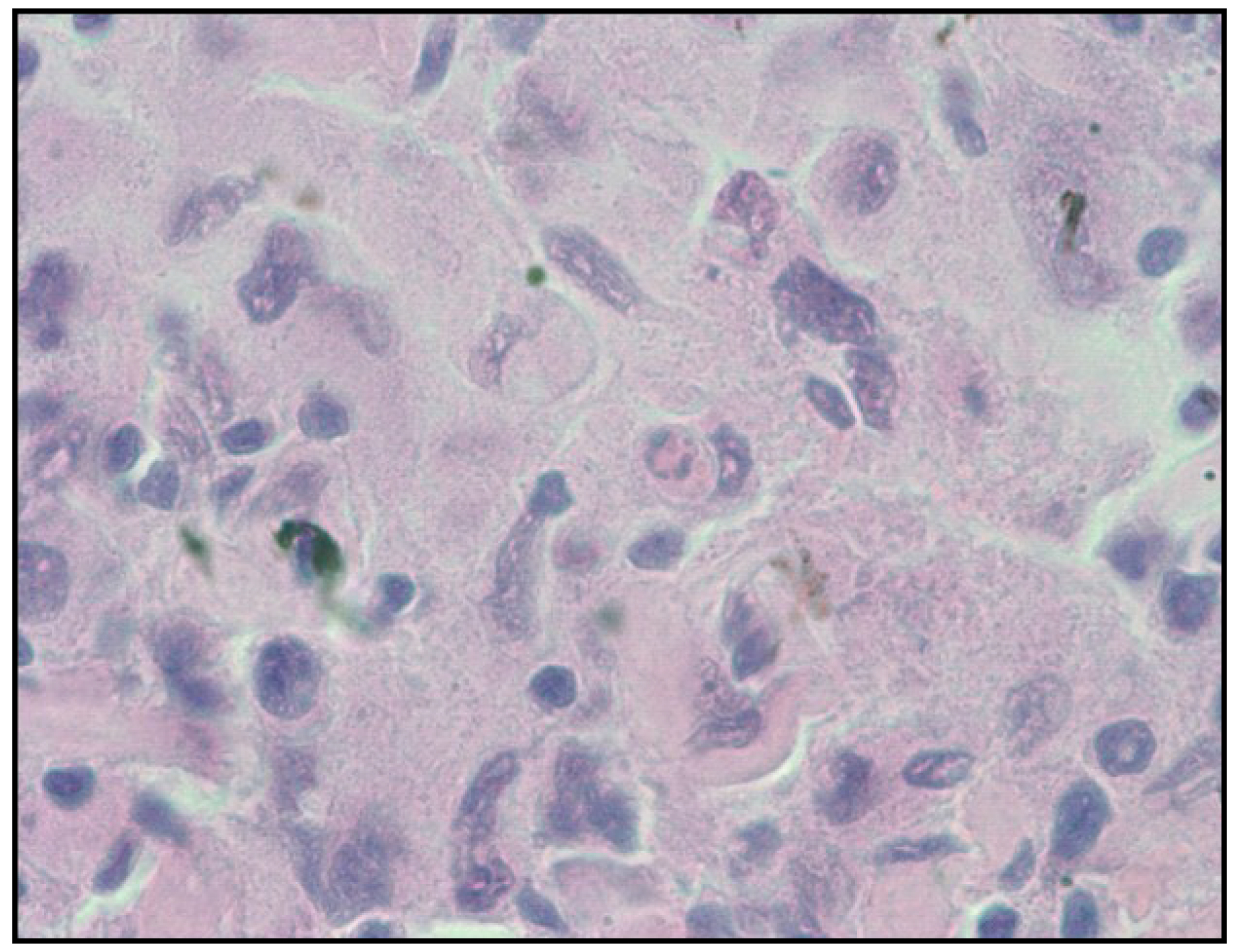
2.2. Histiocytic Sarcoma
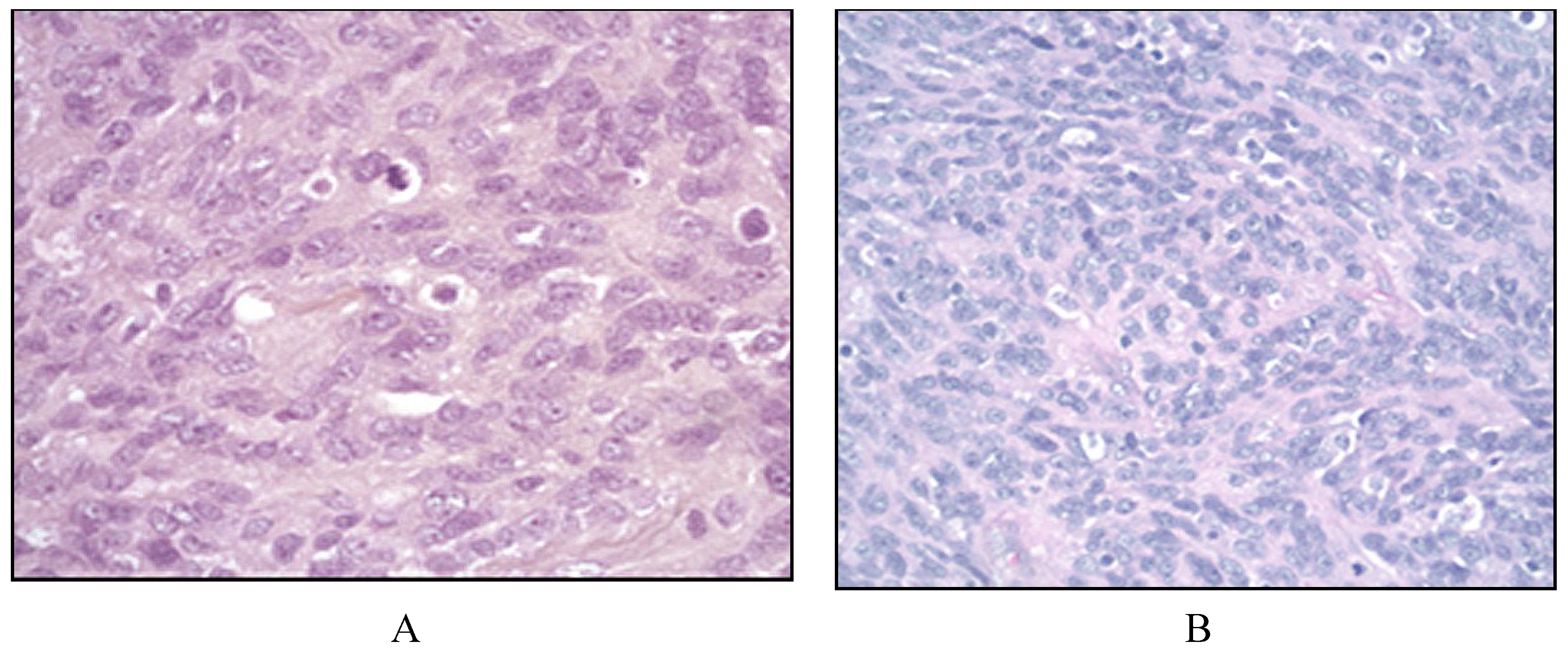
2.3. Follicular Dendritic Cell Sarcoma
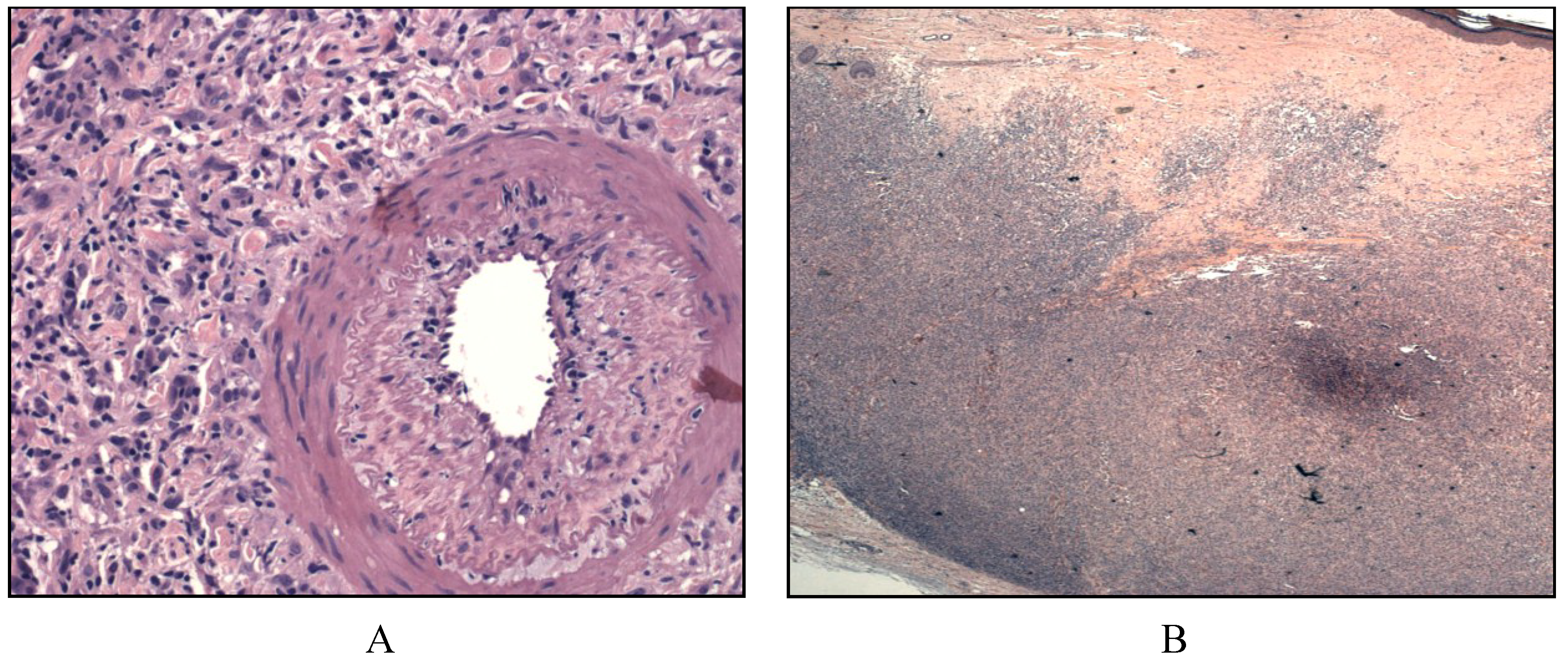
2.4. Interdigitating Dendritic Cell Sarcoma
2.5. Indeterminate Dendritic Cell Sarcoma
2.6. Fibroblastic Reticular Cell Tumor
3. Discussion
3.1. Langerhans Cell Histiocytosis

3.2. Histiocytic Sarcoma

3.3. Follicular Dendritic Cell Sarcoma

3.4. Interdigitating Dendritic Cell Sarcoma

3.5. Indeterminate Dendritic Cell Sarcoma

3.6. Fibroblastic Reticular Cell Tumor
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Shao, H.; Xi, L.; Raffeld, M.; Feldman, A.L.; Ketterling, R.P.; Knudson, R.; Rodriguez-Canales, J.; Hanson, J.; Pittaluga, S.; Jaffe, E.S. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: A study of seven cases. Mod. Pathol. 2011, 24, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; Lyon, France, 2008. [Google Scholar]
- Newman, B.; Hu, W.; Nigro, K.; Gilliam, A.C. Aggressive histiocytic disorders that can involve the skin. J. Am. Acad. Dermatol. 2007, 56, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Pileri, S.A.; Grogan, T.M.; Harris, N.L.; Banks, P.; Campo, E.; Chan, J.K.; Favera, R.D.; Delsol, G.; de Wolf-Peeters, C.; Falini, B.; et al. Tumours of histiocytes and accessory dendritic cells: An immunohistochemical approach to classification from the international lymphoma study group based on 61 cases. Histopathology 2002, 41, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Girschikofsky, M.; Arico, M.; Castillo, D.; Chu, A.; Doberauer, C.; Fichter, J.; Haroche, J.; Kaltsas, G.A.; Makras, P.; Marzano, A.V.; et al. Management of adult patients with Langerhans cell histiocytosis: Recommendations from an expert panel on behalf of EURO-HISTIO-NET. Orphanet J. Rare Dis. 2013. [Google Scholar] [CrossRef]
- Phillips, M.; Allen, C.; Gerson, P.; McClain, K. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr. Blood Cancer 2009, 52, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Pritzker, M.S. Electron microscopic study of reticulohistiocytoma. An unusual case of congenital, self-healing reticulohistiocytosis. Arch. Dermatol. 1973, 107, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Chikwava, K.; Jaffe, R. Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr. Dev. Pathol. 2004, 7, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wheller, L.; Carman, N.; Butler, G. Unilesional self-limited langerhans cell histiocytosis: A case report and review of the literature. J. Cutan. Pathol. 2013, 40, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.L.; Paller, A.S.; Haut, P.R.; Mancini, A.J. Langerhans cell histiocytosis presenting in the neonatal period: A retrospective case series. Arch. Pediatr. Adolesc. Med. 2001, 155, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Munn, S.; Chu, A.C. Langerhans cell histiocytosis of the skin. Hematol. Oncol. Clin. North. Am. 1998, 12, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Holden, C.A.; Broadbent, V.; Atherton, D.J. Histiocytosis X presenting as intertrigo and responding to topical nitrogen mustard. Clin. Exp. Dermatol. 1986, 11, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Taverna, J.A.; Stefanato, C.M.; Wax, F.D.; Demierre, M.F. Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J. Am. Acad. Dermatol. 2006, 54, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.A.; Aughenbaugh, W.; Sharata, H. Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch. Dermatol. 2008, 144, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Lichtenwald, D.J.; Jakubovic, H.R.; Rosenthal, D. Primary cutaneous Langerhans cell histiocytosis in an adult. Arch. Dermatol. 1991, 127, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Imanaka, A.; Tarutani, M.; Itoh, H.; Kira, M.; Itami, S. Langerhans cell histiocytosis involving the skin of an elderly woman: A satisfactory remission with oral prednisolone alone. J. Dermatol. 2004, 31, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Schiltz, C.; Korman, N. Langerhans cell histiocytosis. J. Cutan. Med. Surg. 2012, 16, 45–49. [Google Scholar] [PubMed]
- McLelland, J.; Broadbent, V.; Yeomans, E.; Malone, M.; Pritchard, J. Langerhans cell histiocytosis: The case for conservative treatment. Arch. Dis. Child. 1990, 65, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Stockschlaeder, M.; Sucker, C. Adult Langerhans cell histiocytosis. Eur. J. Haematol. 2006, 76, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Minkov, M.; Grois, N.; McClain, K.; Rodriguez-Galindo, C.; Simonitsch-Klupp, I.; Visser, J.; Wieitzman, S.; Whitlock, J.; Windebank, K. Langerhans cell histiocytosis: Histiocyte society evaluation and treatment guidelines. Available online: https://www.histiocytesociety.org/document.doc?id=290 (accessed on 23 June 2013).
- Gadner, H.; Grois, N.; Potschger, U.; Minkov, M.; Arico, M.; Braier, J.; Broadbent, V.; Donadieu, J.; Henter, J.I.; McCarter, R.; et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 2008, 111, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Gadner, H.; Minkov, M.; Grois, N.; Potschger, U.; Thiem, E.; Arico, M.; Astigarraga, I.; Braier, J.; Donadieu, J.; Henter, J.I.; et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood 2013, 121, 5006–5014. [Google Scholar] [CrossRef] [PubMed]
- Badalian-Very, G.; Vergilio, J.A.; Degar, B.A.; MacConaill, L.E.; Brandner, B.; Calicchio, M.L.; Kuo, F.C.; Ligon, A.H.; Stevenson, K.E.; Kehoe, S.M.; et al. Recurrent braf mutations in Langerhans cell histiocytosis. Blood 2010, 116, 1919–1923. [Google Scholar] [CrossRef] [PubMed]
- Bernard, F.; Thomas, C.; Bertrand, Y.; Munzer, M.; Landman Parker, J.; Ouache, M.; Colin, V.M.; Perel, Y.; Chastagner, P.; Vermylen, C.; et al. Multi-centre pilot study of 2-chloro-deoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur. J. Cancer 2005, 41, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Haroche, J.; Cohen-Aubart, F.; Emile, J.F.; Arnaud, L.; Maksud, P.; Charlotte, F.; Cluzel, P.; Drier, A.; Hervier, B.; Benameur, N.; et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013, 121, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Jaffe, E.S.; Fletcher, C.D. Extranodal histiocytic sarcoma: Clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am. J. Surg. Pathol. 2004, 28, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Meck, J.; Cheson, B.D.; Ozdemirli, M. Histiocytic sarcoma transdifferentiated from follicular lymphoma presenting as a cutaneous tumor. J. Cutan. Pathol. 2011, 38, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Arber, D.A.; Pittaluga, S.; Martinez, A.; Burke, J.S.; Raffeld, M.; Camos, M.; Warnke, R.; Jaffe, E.S. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: Evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008, 111, 5433–5439. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.A.; Abbondanzo, S.L.; Barekman, C.L.; Andriko, J.W.; Miettinen, M.; Aguilera, N.S. Histiocytic sarcoma: A study of five cases including the histiocyte marker CD163. Mod. Pathol. 2005, 18, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Gianotti, F.; Caputo, R. Histiocytic syndromes: A review. J. Am. Acad. Dermatol. 1985, 13, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Yamakawa, M.; Nakamura, S.; van Heerde, P.; Miettinen, M.; Shek, T.W.; Myhre Jensen, O.; Rousselet, M.C.; Tefferi, A. Follicular dendritic cell sarcoma and interdigitating reticulum cell sarcoma: A review. Am. J. Hematol. 1998, 59, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Tew, J.G.; Kosco, M.H.; Burton, G.F.; Szakal, A.K. Follicular dendritic cells as accessory cells. Immunol. Rev. 1990, 117, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qin, D.; Burton, G.F.; Szakal, A.K.; Tew, J.G. Follicular dendritic cell-derived antigen and accessory activity in initiation of memory IGg responses in vitro. J. Immunol. 1996, 157, 3404–3411. [Google Scholar] [PubMed]
- Pauwels, P.; dal Cin, P.; Vlasveld, L.T.; Aleva, R.M.; van Erp, W.F.; Jones, D. A chromosomal abnormality in hyaline vascular Castlemanʼs disease: Evidence for clonal proliferation of dysplastic stromal cells. Am. J. Surg. Pathol. 2000, 24, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Cokelaere, K.; Debiec-Rychter, M.; de Wolf-Peeters, C.; Hagemeijer, A.; Sciot, R. Hyaline vascular Castleman’s disease with hmgic rearrangement in follicular dendritic cells: Molecular evidence of mesenchymal tumorigenesis. Am. J. Surg. Pathol. 2002, 26, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chang, K.C.; Abruzzo, L.V.; Lai, R.; Younes, A.; Jones, D. Epidermal growth factor receptor expression in follicular dendritic cells: A shared feature of follicular dendritic cell sarcoma and Castlemanʼs disease. Hum. Pathol. 2003, 34, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Schwartz, E.J.; West, R.B.; Warnke, R.A.; Arber, D.A.; Natkunam, Y. Expression of CD163 (hemoglobin scavenger receptor) in normal tissues, lymphomas, carcinomas, and sarcomas is largely restricted to the monocyte/macrophage lineage. Am. J. Surg. Pathol. 2005, 29, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Uzunaslan, D.; Ozguroglu, M.; Senocak, M.; Tuzuner, N. Dendritic cell sarcoma: A pooled analysis including 462 cases with presentation of our case series. Crit. Rev. Oncol. Hematol. 2013, 88, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Fletcher, C.D.; Nayler, S.J.; Cooper, K. Follicular dendritic cell sarcoma. Clinicopathologic analysis of 17 cases suggesting a malignant potential higher than currently recognized. Cancer 1997, 79, 294–313. [Google Scholar] [CrossRef] [PubMed]
- Shia, J.; Chen, W.; Tang, L.H.; Carlson, D.L.; Qin, J.; Guillem, J.G.; Nobrega, J.; Wong, W.D.; Klimstra, D.S. Extranodal follicular dendritic cell sarcoma: Clinical, pathologic, and histogenetic characteristics of an underrecognized disease entity. Virchows Arch. 2006, 449, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.M.; Shinohara, E.T. Interdigitating and follicular dendritic cell sarcomas: A SEER analysis. Am. J. Clin. Oncol. 2013, 36, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Ylagan, L.R.; Bartlett, N.L.; Kraus, M. Interdigitating dendritic reticulum cell tumor of lymph nodes: Case report with differential diagnostic considerations. Diagn. Cytopathol. 2003, 28, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, Y.H.; Guo, Z.J.; Qiu, T.; Guo, L.; Yang, H.Y.; Zhang, X.; Zhao, X.M.; Su, Q. Clinicopathological features and prognosis assessment of extranodal follicular dendritic cell sarcoma. World J. Gastroenterol. 2010, 16, 2504–2519. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.O.; Thompson, M.A.; Admirand, J.H.; Fayad, L.E.; Rodriguez, A.M.; Romaguera, J.E.; Hagemeister, F.B.; Pro, B. Follicular dendritic cell sarcoma: A report of 14 cases and a review of the literature. Am. J. Hematol. 2007, 82, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Pack, M.; Inaba, K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol. Rev. 1997, 156, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Takahashi, T.; Saitoh, C.; Watarai, K.; Wade, K.; Tajim, K.; Harada, Y.; Hiroshima, Y.; Yamakawa, M. Interdigitating cell sarcoma: A report of an autopsy case and literature review. J. Clin. Exp. Hematopathol. 2005, 45, 37–44. [Google Scholar] [CrossRef]
- Imai, Y.; Yamakawa, M.; Kasajima, T. The lymphocyte-dendritic cell system. Histol. Histopathol. 1998, 13, 469–510. [Google Scholar] [PubMed]
- Steinman, R.M. Some interfaces of dendritic cell biology. APMIS 2003, 111, 675–697. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.P.; Chen, Y.; Chan, V.S.; Tam, P.K.; Lin, C.L. Dendritic cells: Sentinels against pathogens. Histol. Histopathol. 2004, 19, 317–324. [Google Scholar] [PubMed]
- Fraser, C.R.; Wang, W.; Gomez, M.; Zhang, T.; Mathew, S.; Furman, R.R.; Knowles, D.M.; Orazi, A.; Tam, W. Transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma to interdigitating dendritic cell sarcoma: Evidence for transdifferentiation of the lymphoma clone. Am. J. Clin. Pathol. 2009, 132, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, E.M.; Tsokos, M.; Derringer, G.A.; Neuhauser, T.S.; Arciero, C.; Andriko, J.A. Interdigitating dendritic cell sarcoma. A report of four cases and review of the literature. Am. J. Clin. Pathol. 2001, 115, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Dong, N.Z.; Wu, D.P.; Xue, S.L. Interdigitating dendritic cell sarcoma presenting simultaneously with acute myelomonocytic leukemia: Report of a rare case and literature review. Int. J. Hematol. 2013, 97, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Christensen, T.; O’Hara, C.J. Metastatic interdigitating dendritic cell sarcoma masquerading as a skin primary tumor: A case report and review of the literature. Am. J. Dermatopathol. 2009, 31, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, C.; Lei, L.; Yan, X.; Wang, B.; Liu, X.; Yv, L.; Lv, Y. Interdigitating dendritic cell sarcoma involving bone marrow in a liver transplant recipient. Transplant. Proc. 2010, 42, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.; Eisinger, M.; Lee, J.S.; Takezaki, S.; Kung, P.C.; Edelson, R.L. Immunoelectron microscopic identification of langerhans cells using a new antigenic marker. J. Invest. Dermatol. 1982, 78, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Lenz, A.; Glassel, H.; Stossel, H.; Stanzl, U.; Majdic, O.; Fritsch, P.; Schuler, G. Cultured human Langerhans cells resemble lymphoid dendritic cells in phenotype and function. J. Invest. Dermatol. 1989, 93, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.B.; Wormmeester, J.; Krieg, S.R.; Peters, P.J.; Vogels, I.M.; Kapsenberg, M.L.; Bos, J.D. Human epidermal Langerhans cells undergo profound morphologic and phenotypical changes during in vitro culture. J. Invest. Dermatol. 1990, 94, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Weber, L.; Scharffetter-Kochanek, K.; Weiss, J.M. Solitary cutaneous dendritic cell tumor in a child: Role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J. Am. Acad. Dermatol. 2005, 53, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Ratzinger, G.; Burgdorf, W.H.; Metze, D.; Zelger, B.G.; Zelger, B. Indeterminate cell histiocytosis: Fact or fiction? J. Cutan. Pathol. 2005, 32, 552–560. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fujiwara, K.; Punwaney, J.; DiGregorio, F.; Bostrom, P.; El-Hoshy, K.; Aronson, P.J.; Schoenfeld, R.J. Post-scabetic nodules: A lymphohistiocytic reaction rich in indeterminate cells. J. Dermatol. 2000, 27, 181–194. [Google Scholar] [PubMed]
- Wollenberg, A.; Burgdorf, W.H.; Schaller, M.; Sander, C. Long-lasting “christmas tree rash” in an adolescent: Isotopic response of indeterminate cell histiocytosis in pityriasis rosea? Acta Derm. Venereol. 2002, 82, 288–291. [Google Scholar] [CrossRef]
- Vasef, M.A.; Zaatari, G.S.; Chan, W.C.; Sun, N.C.; Weiss, L.M.; Brynes, R.K. Dendritic cell tumors associated with low-grade B-cell malignancies. Report of three cases. Am. J. Clin. Pathol. 1995, 104, 696–701. [Google Scholar] [PubMed]
- Segal, G.H.; Mesa, M.V.; Fishleder, A.J.; Stoler, M.H.; Weick, J.K.; Lichtin, A.E.; Tubbs, R.R. Precursor Langerhans cell histiocytosis. An unusual histiocytic proliferation in a patient with persistent non-Hodgkin lymphoma and terminal acute monocytic leukemia. Cancer 1992, 70, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Ferran, M.; Toll, A.; Gilaberte, M.; Barranco, C.; Lloreta, J.; Pujol, R.M. Acquired mucosal indeterminate cell histiocytoma. Pediatr. Dermatol. 2007, 24, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Morgan, M.B. Cutaneous indeterminate cell histiocytosis: A new spindle cell variant resembling dendritic cell sarcoma. J. Cutan. Pathol. 2001, 28, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Martel, M.; Sarli, D.; Colecchia, M.; Coppa, J.; Romito, R.; Schiavo, M.; Mazzaferro, V.; Rosai, J. Fibroblastic reticular cell tumor of the spleen: Report of a case and review of the entity. Hum. Pathol. 2003, 34, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Andriko, J.W.; Kaldjian, E.P.; Tsokos, M.; Abbondanzo, S.L.; Jaffe, E.S. Reticulum cell neoplasms of lymph nodes: A clinicopathologic study of 11 cases with recognition of a new subtype derived from fibroblastic reticular cells. Am. J. Surg. Pathol. 1998, 22, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalia, S.; Jaglal, M.; Chervenick, P.; Cualing, H.; Sokol, L. Clinicopathologic Characteristics and Outcomes of Histiocytic and Dendritic Cell Neoplasms: The Moffitt Cancer Center Experience Over the Last Twenty Five Years. Cancers 2014, 6, 2275-2295. https://doi.org/10.3390/cancers6042275
Dalia S, Jaglal M, Chervenick P, Cualing H, Sokol L. Clinicopathologic Characteristics and Outcomes of Histiocytic and Dendritic Cell Neoplasms: The Moffitt Cancer Center Experience Over the Last Twenty Five Years. Cancers. 2014; 6(4):2275-2295. https://doi.org/10.3390/cancers6042275
Chicago/Turabian StyleDalia, Samir, Michael Jaglal, Paul Chervenick, Hernani Cualing, and Lubomir Sokol. 2014. "Clinicopathologic Characteristics and Outcomes of Histiocytic and Dendritic Cell Neoplasms: The Moffitt Cancer Center Experience Over the Last Twenty Five Years" Cancers 6, no. 4: 2275-2295. https://doi.org/10.3390/cancers6042275
APA StyleDalia, S., Jaglal, M., Chervenick, P., Cualing, H., & Sokol, L. (2014). Clinicopathologic Characteristics and Outcomes of Histiocytic and Dendritic Cell Neoplasms: The Moffitt Cancer Center Experience Over the Last Twenty Five Years. Cancers, 6(4), 2275-2295. https://doi.org/10.3390/cancers6042275