
cMET in NSCLC: Can We Cut off the Head of the Hydra? From the Pathway to the Resistance




{kind=link}
Abstract
:1. Introduction
2. cMET and HGF Structure
3. The cMET Pathway Disassembled
3.1. Ligand-Dependent Activation
3.1.1. MAPK Cascades
3.1.2. PI3K-Akt
3.1.3. STAT3
3.1.4. NF-κB
3.2. Ligand Independent Activation
3.2.1. α5β1-Integrines-FAK
3.2.2. Sema4D
3.3. cMET Internalization
3.4. cMET Shedding
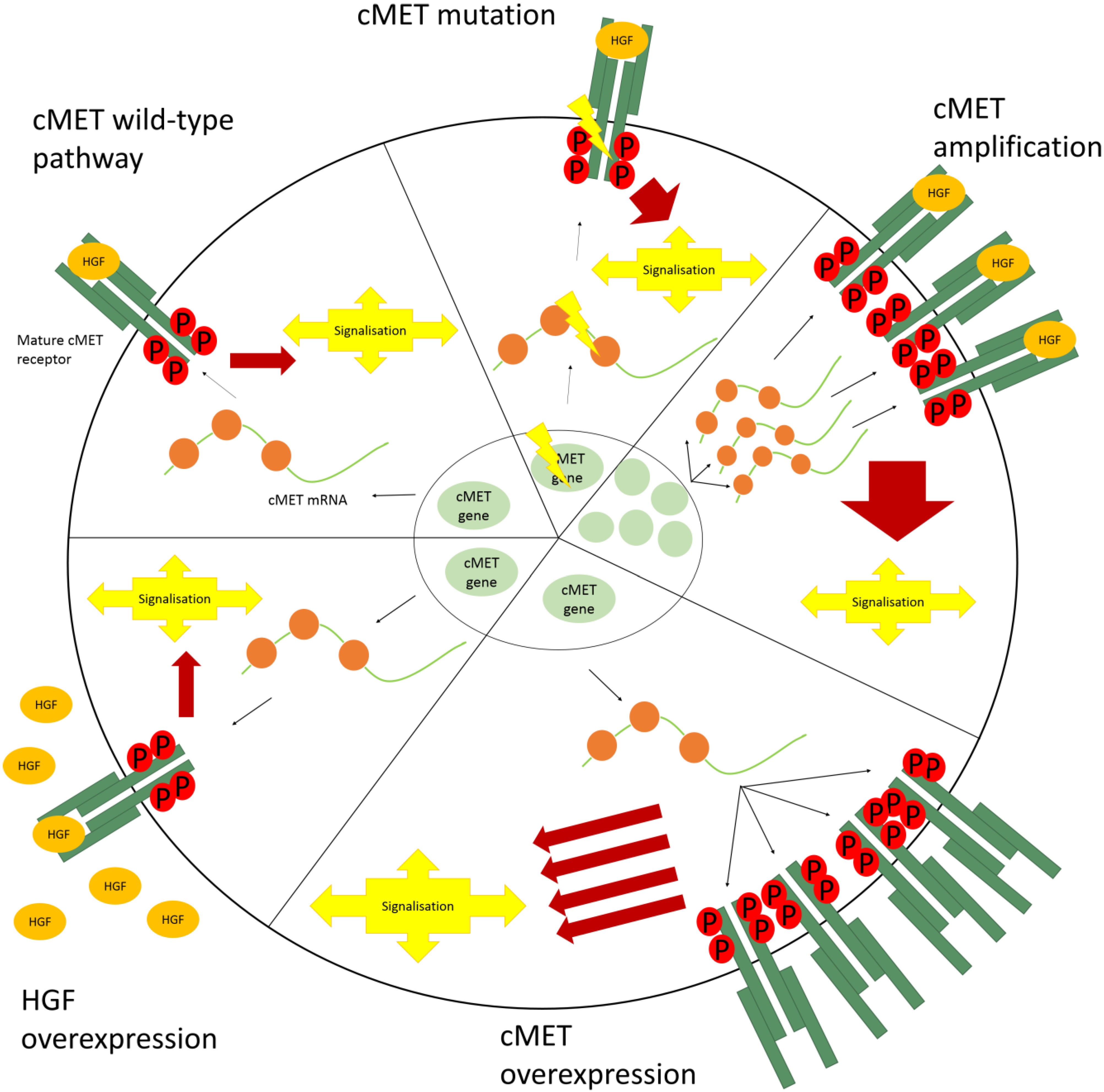
4. Aberrant cMET Signaling
4.1. cMET Mutations
4.2. Amplification
4.3. Overexpression

4.4. HGF Overexpression
5. cMET as a Resistance Mechanism in the Treatment of NSCLC
5.1. cMET and Ionizing Radiation
5.2. cMET and Chemotherapy
5.3. cMET and Hypoxia
5.4. cMET and EGFR-Inhibition
6. Discussion
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Achim, C.L.; Katyal, S.; Wiley, C.; Shiratori, M.; Wang, G.; Oshika, E.; Petersen, B.E.; Li, J.M.; Michalopoulos, G.K. Expression of HGF and cMET in the developing and adult brain. Dev. Brain Res. 1997, 102, 299–303. [Google Scholar] [CrossRef]
- Conway, K.P.; Price, P.E.; Harding, K.G.; Jiang, W.G. The molecular and clinical impact of hepatocyte growth factor, its receptor, activators, and inhibitors in wound healing. Wound Repair Regen. 2006, 14, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef] [PubMed]
- Montesano, R.; Soriano, J.V.; Malinda, K.M.; Ponce, M.L.; Bafico, A.; Kleinman, H.K.; Bottaro, D.P.; Aaronson, S. A Differential effects of hepatocyte growth factor isoforms on epithelial and endothelial tubulogenesis. Cell Growth Differ. 1998, 9, 355–365. [Google Scholar] [PubMed]
- Ding, S.; Merkulova-rainon, T.; Han, Z.C. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 2003, 101, 4816–4822. [Google Scholar] [CrossRef] [PubMed]
- Kajiya, K.; Hirakawa, S.; Ma, B.; Drinnenberg, I.; Detmar, M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005, 24, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Stewart, F. Roles of mesenchymal-epithelial interactions and hepatocyte growth factor-scatter factor (HGF-SF) in placental development. Rev. Reprod. 1996, 1, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Santos, O.; Barros, E.; Yang, X.-M.; Matsumoto, K.; Nakamura, T.; Park, M.; Nigam, S. Involvement of hepatocyte growth factor in kidney development. Dev. Biol. 1994, 163, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Komada, M.; Hatsuzawa, K.; Shibamoto, S.; Ito, F.; Nakayama, K.; Kitamura, N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett. 1993, 328, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tempest, P.; Stratton, M.; Cooper, C. Structure of the met protein and variation of met protein kinase activity among human tumour cell lines. Br. J. Cancer 1988, 58, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Flavia, M.; Renzo, D.I.; Ferracini, R.; Chiado-piat, L.; Comoglio, P.M. p145, a protein with associated tyrosine kinase activity in human gastric carcinoma cell line. Mol. Cell Biol. 1988, 8, 3510–3517. [Google Scholar] [PubMed]
- Gherardi, E.; Youles, M.E.; Miguel, R.N.; Blundell, T.L.; Iamele, L.; Gough, J.; Bandyopadhyay, A.; Hartmann, G.; Butler, P.J.G. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 2003, 100, 12039–12044. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Giordano, S.; Graxiani, A.; Panayotou, G.; Comoglio, P.M.Y.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Castren, E.; Odenthal, M.; vande Woude, G.F.; Ishii, T.; Dienes, H.P.; Lindholm, D.; Schirmacher, P. Expression and functional interaction of hepatocyte growth factor—Scatter factor and its receptor. J. Cell Biol. 1994, 126, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, E.; Meyer, D.; Weidner, K.M.; Birchmeier, C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J. Cell Biol. 1993, 123, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am. J. Pathol. 1993, 143, 949–958. [Google Scholar] [PubMed]
- Owen, K.A.; Qiu, D.; Alves, J.; Schumacher, A.M.; Kilpatrick, L.M.; Li, J.; Harris, J.L.; Ellis, V. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. Biochem. J. 2010, 426, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, K.; Inoue, H.; Hagiya, M.; Shimizu, S.; Nose, T.; Shimohigasho, Y.; Nakamura, T. Hairpin loop and second kringle domain are essential sites heparin binding and biological activity of hepatocyte growth factor. J. Biol. Chem. 1994, 269, 1131–1136. [Google Scholar] [PubMed]
- Sakata, H.; Stahl, S.J.; Taylor, W.G.; Rosenberg, J.M.; Sakaguchi, K.; Wingfield, P.T.; Rubin, J.S. Heparin binding and oligomerization of hepatocyte growth factor/scatter factor isoforms. Heparan Sulfate glycosaminoglycan requirement for met binding and signaling. J. Biol. Chem. 1997, 272, 9457–9463. [Google Scholar] [CrossRef] [PubMed]
- Fixman, E.; Fournier, T.; Kamikura, D.; Naujokas, M.; Park, M. Pathways downstream of Shc and Grb2 are required for cell transformation by the Tpr-Met oncoprotein. J. Biol. Chem. 1996, 271, 13116–13122. [Google Scholar] [CrossRef] [PubMed]
- Weinder, K.M.; di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Orian-rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-Actin. Mol. Biol. Cell 2007, 18, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Yart, A.; Dance, M.; Perret, B.; Salles, J.-P.; Raynal, P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J. Biol. Chem. 2005, 280, 5350–5360. [Google Scholar] [CrossRef] [PubMed]
- Graziani, A.; Gramaglia, D.; dalla Zonca, P.; Comoglio, P.M. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J. Biol. Chem. 1993, 268, 9165–9168. [Google Scholar] [PubMed]
- Xiao, G.H.; Jeffers, M.; Bellacosa, A.; Mitsuuchi, Y.; vande Woude, G.F.; Testa, J.R. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Sabatini, D.; Efeyan, A. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2012, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3β in cell survival and NF-kB activation. Nature 2000, 406, 2–6. [Google Scholar] [CrossRef]
- Del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-Induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Patané, S.; Porras, A.; Dono, R.; Maina, F. Met acts on Mdm2 via mTOR to signal cell survival during development. Development 2007, 134, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Wang, L.-M.; Jove, R.; vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Morotti, A.; Ponzetto, C. Activation of NF-kB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol. Cell. Biol. 2002, 22, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Gao, M.; Meng, Q.; Laterra, J.J.; Symons, M.H.; Coniglio, S.; Pestell, R.G.; Goldberg, I.D.; Rosen, E.M. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene 2005, 24, 1749–1766. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kobayashi, R.; Bishop, J.M. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 8425–8430. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Matsubara, D.; Goto, A.; Ota, S.; Sachiko, O.; Ishikawa, S.; Aburatani, H.; Miyazawa, K.; Fukayama, M.; Niki, T. Constitutive activation of c-Met is correlated with c-Met overexpression and dependent on cell-matrix adhesion in lung adenocarcinoma cell lines. Cancer Sci. 2008, 99, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand independent activation of c-Met by fibronectin and α5 β1—Integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.Y.; Meens, J.A.; Schick, C.; Organ, S.L.; Qiao, H.; Tremblay, E.A.; Schaeffer, E.; Uniyal, S.; Chan, B.M.C.; Elliott, B.E. Src and FAK mediate cell-matrix adhesion-dependent activation of Met during transformation of breast epithelial cells. J. Cell. Biochem. 2009, 107, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Basile, J.R.; Afkhami, T.; Gutkind, J.S. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol. Cell. Biol. 2005, 25, 6889–6898. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Corso, S.; Conrotto, P.; Artigiani, S.; Gilestro, G.; Barberis, D.; Tamagnone, L.; Comoglio, P.M. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 2002, 4, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Basile, J.R.; Castilho, R.M.; Williams, V.P.; Gutkind, J.S. Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 9017–9022. [Google Scholar] [CrossRef] [PubMed]
- Conrotto, P.; Corso, S.; Gamberini, S.; Comoglio, P.M.; Giordano, S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene 2004, 23, 5131–5137. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Holtrich, U.; Gaetje, R.; Gehrmann, M.; Engels, K.; von Minckwitz, G.; Loibl, S.; Diallo-Danebrock, R.; Ruckhäberle, E.; Metzler, D.; et al. Poor outcome in estrogen receptor-positive breast cancers predicted by loss of plexin B1. Clin. Cancer Res. 2007, 13, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Karn, T.; Ruckhäberle, E.; Hanker, L.; Metzler, D.; Müller, V.; Solbach, C.; Ahr, A.; Gätje, R.; Holtrich, U.; et al. Loss of Plexin B1 is highly prognostic in low proliferating ER positive breast cancers—Results of a large scale microarray analysis. Eur. J. Cancer 2009, 45, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Soong, J.; Chen, Y.; Shustef, E.M.; Scott, G.A. Sema4D, the ligand for Plexin B1, suppresses c-Met activation and migration and promotes melanocyte survival and growth. J. Investig. Dermatol. 2012, 132, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Parker, J. c-MET signalling: Spatio-temporal decisions. Cell Cycle 2005, 4, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Parachoniak, C.A.; Luo, Y.; Abella, J.V.; Keen, J.H.; Park, M. GGA3 functions as a switch to promote Met receptor recycling, essential for sustained ERK and cell migration. Dev. Cell 2011, 20, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Urbe, S.; vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Ceresa, B.P.; Kao, A.W.; Santeler, S.R.; Pessin, J.E. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol. Cell. Biol. 1998, 18, 3862–3870. [Google Scholar] [PubMed]
- Kermorgant, S.; Parker, P.J. Receptor trafficking controls weak signal delivery: A strategy used by c-Met for STAT3 nuclear accumulation. J. Cell Biol. 2008, 182, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Ménard, L.; Parker, P.J.; Kermorgant, S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 2014, 5, 3907. [Google Scholar] [PubMed]
- Foveau, B.; Ancot, F.; Leroy, C.; Petrelli, A.; Reiss, K.; Vingtdeux, V.; Giordano, S.; Fafeur, V.; Tulasne, D. Down-regulation of the Met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol. Biol. Cell 2009, 20, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Kopitz, C.; Gerg, M.; Bandapalli, O.R.; Ister, D.; Pennington, C.J.; Hauser, S.; Flechsig, C.; Krell, H.-W.; Antolovic, D.; Brew, K.; et al. Tissue inhibitor of metalloproteinases-1 promotes liver metastasis by induction of hepatocyte growth factor signaling. Cancer Res. 2007, 67, 8615–8623. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Kanteti, R.; Duke-cohan, J.S.; Loganathan, S.; Liu, W.; Ma, P.C.; Sattler, M.; Singleton, P.A.; Ramnath, N.; Innocenti, F.; et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin. Cancer Res. 2009, 15, 5714–5723. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar] [PubMed]
- Jagadeeswaran, R.; Jagadeeswaran, S.; Bindokas, V.P.; Salgia, R. Activation of HGF/c-Met pathway contributes to the reactive oxygen species generation and motility of small cell lung cancer cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1488–L1494. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Fletcher, L.B.; Wang, E.Q.; Yang, W.F.; Rutenberg-Schoenberg, M.L.; Beadling, C.; Mori, M.; Heinrich, M.C.; Deininger, M.W.; Druker, B.J.; et al. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010, 70, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Han, S.; Cho, H.; Jennings, B.; Gerrard, B.; Dean, M.; Schmidt, L.; Zbar, B.; vande Woude, G.F. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene 2000, 19, 4947–4953. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Timofeevski, S.L.; McTigue, M.A.; Ryan, K.; Cui, J.; Zou, H.Y.; Zhu, J.X.; Chau, F.; Alton, G.; Karlicek, S.; Christensen, J.G.; et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry 2009, 48, 5339–5349. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Schmidt, L.; Nakaigawa, N.; Webb, C.P.; Weirich, G.; Kishida, T.; Zbar, B.; vande woude, G.F. Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; vande Woude, G.F. Activating mutations in the Met receptor overcome the requirement for autophosphorylation of tyrosines crucial for wild type signaling. Oncogene 1999, 18, 5120–5125. [Google Scholar] [CrossRef] [PubMed]
- Chiara, F.; Michieli, P.; Pugliese, L.; Comoglio, P.M. Mutations in the met oncogene unveil a “dual switch” mechanism controlling tyrosine kinase activity. J. Biol. Chem. 2003, 278, 29352–29358. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Mok, T.S.; Chen, Z.-H.; Guo, A.-L.; Zhang, X.-C.; Su, J.; Wu, Y.-L. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol. Oncol. Res. 2009, 15, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, Y.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207. [Google Scholar] [PubMed]
- Sun, W.; Song, L.; Ai, T.; Zhang, Y.; Gao, Y.; Cui, J. Prognostic value of MET, cyclin D1 and MET gene copy number in non-small cell lung cancer. J. Biomed. Res. 2013, 27, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Streubel, B.; Berghoff, A.S.; Hainfellner, J.A.; von Deimling, A.; Widhalm, G.; Dieckmann, K.; Wöhrer, A.; Hackl, M.; Zielinski, C.; et al. Amplification and overexpression of CMET is a common event in brain metastases of non-small cell lung cancer. Histopathology 2014, 65, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Tsuta, K.; Kozu, Y.; Mimae, T.; Yoshida, A.; Kohno, T.; Sekine, I.; Tamura, T.; Asamura, H.; Furuta, K.; Tsuda, H. c-MET/phospho-MET protein expression and MET gene copy number in non-small cell lung carcinomas. J. Thorac. Oncol. 2012, 7, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Elia, G.; Ren, Y.; Lorenzoni, P.; Zarnegar, R.; Burger, M.M.; Rusciano, D. Mechanisms regulating c-met overexpression in liver-metastatic B16-LS9 melanoma cells. J. Cell. Biochem. 2001, 81, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Puszyk, W.; Dong, H.J.; Liu, C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef]
- Garofalo, M.; Romano, G.; di Leva, G.; Nuovo, G.; Jeon, Y.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase—Altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2011, 18, 74–82. [Google Scholar] [PubMed]
- Rahimi, N.; Tremblay, E.; Mcadam, L.; Park, M.; Schwall, R.; Eliiott, B.; Kl, O.; Canada, N.R.; Victoria, R. Identification of a hepatocyte growth factor autocrine loop in a murine mammary carcinoma. Cell Growth Differ. 1996, 7, 263–270. [Google Scholar] [PubMed]
- Trovato, M.; Vitarelli, E.; Grosso, M.; Alesci, S.; Benvenga, S.; Trimarchi, F.; Barresi, G. Immunohistochemical expression of HGF, c-MET and transcription factor STAT3 in colorectal tumors. Eur. J. Histochem. 2004, 48, 291–297. [Google Scholar] [PubMed]
- Masuya, D.; Huang, C.; Liu, D.; Nakashima, T.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br. J. Cancer 2004, 90, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Cascone, T.; Cortez, M.A.; Amini, A.; Evans, J.; Komaki, R.U.; Heymach, J.V.; Welsh, J.W. Modulation of c-Met signaling and cellular sensitivity to radiation: Potential implications for therapy. Cancer 2013, 119, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.-W.; Mizumoto, K.; Inadome, N.; Nagai, E.; Sato, N.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation stimulates HGF receptor/c-Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. Int. J. Cancer 2003, 104, 542–549. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment—Tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, J.N.; Redmond, K.M.; Schettino, G.; Prise, K.M. DNA double strand break repair: A radiation perspective. Antioxid. Redox Signal. 2013, 18, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Medová, M.; Aebersold, D.M.; Zimmer, Y. MET inhibition in tumor cells by PHA665752 impairs homologous recombination repair of DNA double strand breaks. Int. J. Cancer 2012, 130, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Sheng-Hua, C.; Yan-Bin, M.; Zhi-An, Z.; Hong, Z.; Dong-Fu, F.; Zhi-Qiang, L.; Xian-Hou, Y. Radiation-enhanced hepatocyte growth factor secretion in malignant glioma cell lines. Surg. Neurol. 2007, 68, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Vertes-George, E.; Hughes, S.J.; Behrns, K.E.; Trevino, J.G. c-Met signaling in the development of tumorigenesis and chemoresistance: Potential applications in pancreatic cancer. World J. Gastroenterol. 2014, 20, 8458–8470. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.K.S.; Zhou, H.Y.; Yam, J.W.P.; Wong, A.S.T. c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia 2010, 12, 128–138. [Google Scholar] [PubMed]
- Chen, J.-T.; Huang, C.-Y.; Chiang, Y.-Y.; Chen, W.-H.; Chiou, S.-H.; Chen, C.-Y.; Chow, K.-C. HGF increases cisplatin resistance via down-regulation of AIF in lung cancer cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Marchion, D.C.; Bicaku, E.; Xiong, Y.; Zgheib, N.B.; al Sawah, E.; Stickles, X.B.; Judson, P.L.; Lopez, A.S.; Cubitt, C.L.; Gonzalez-Bosquet, J.; et al. A novel c-Met inhibitor, MK8033, synergizes with carboplatin plus paclitaxel to inhibit ovarian cancer cell growth. Oncol. Rep. 2013, 29, 2011–2018. [Google Scholar] [PubMed]
- Yashiro, M.; Nishii, T.; Hasegawa, T.; Matsuzaki, T.; Morisaki, T.; Fukuoka, T.; Hirakawa, K. A c-Met inhibitor increases the chemosensitivity of cancer stem cells to the irinotecan in gastric carcinoma. Br. J. Cancer 2013, 1, 1–10. [Google Scholar]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Koga, Y.; Miyazaki, K. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int. J. Cancer 2006, 119, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Taron, M.; Ichinose, Y.; Rosell, R.; Mok, T.; Massuti, B.; Zamora, L.; Mate, J.L.; Manegold, C.; Ono, M.; Queralt, C.; et al. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin. Cancer Res. 2005, 11, 5878–5885. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Benedettini, E.; Sholl, L.M.; Peyton, M.; Reilly, J.; Ware, C.; Davis, L.; Vena, N.; Bailey, D.; Yeap, B.Y.; Fiorentino, M.; et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am. J. Pathol. 2010, 177, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Jänne, P.A.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.A.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2009, 20, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Nanjo, S.; Ishikawa, D.; Sano, T.; Kita, K.; Nakamura, T.; Matsumoto, K.; Suda, K.; et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol. Cancer Ther. 2012, 11, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.-Y.; Zhao, J.; Chen, J.-J.; Ma, S.-N.; Zhou, J.-Y. Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cells. Anticancer Drugs 2013, 24, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.-J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.-M.; et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to to EGFR kinase inhibitors due to MET or AXL activation. Cancer Res. 2014, 74, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; An, S.J.; Chen, Z.H.; Su, J.; Yan, H.H.; Wu, Y.L. MET expression plays differing roles in non-small-cell lung cancer patients with or without EGFR mutation. J. Thorac. Oncol. 2014, 9, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Wynes, M.W.; Singh, S.; Asuncion, B.R.; Ranger-Moore, J.; Konopa, K.; Rzyman, W.; Szostakiewicz, B.; Jassem, J.; Hirsch, F.R. Correlation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 997–1003. [Google Scholar]
- Etnyre, D.; Stone, A.L.; Fong, J.T.; Jacobs, R.J.; Uppada, S.B.; Botting, G.M.; Rajanna, S.; Moravec, D.N.; Shambannagari, M.R.; Crees, Z.; et al. Targeting c-Met in melanoma. Cancer Biol. Ther. 2014, 15, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Der Steen, N.; Pauwels, P.; Gil-Bazo, I.; Castañon, E.; Raez, L.; Cappuzzo, F.; Rolfo, C. cMET in NSCLC: Can We Cut off the Head of the Hydra? From the Pathway to the Resistance. Cancers 2015, 7, 556-573. https://doi.org/10.3390/cancers7020556
Van Der Steen N, Pauwels P, Gil-Bazo I, Castañon E, Raez L, Cappuzzo F, Rolfo C. cMET in NSCLC: Can We Cut off the Head of the Hydra? From the Pathway to the Resistance. Cancers. 2015; 7(2):556-573. https://doi.org/10.3390/cancers7020556
Chicago/Turabian StyleVan Der Steen, Nele, Patrick Pauwels, Ignacio Gil-Bazo, Eduardo Castañon, Luis Raez, Federico Cappuzzo, and Christian Rolfo. 2015. "cMET in NSCLC: Can We Cut off the Head of the Hydra? From the Pathway to the Resistance" Cancers 7, no. 2: 556-573. https://doi.org/10.3390/cancers7020556
APA StyleVan Der Steen, N., Pauwels, P., Gil-Bazo, I., Castañon, E., Raez, L., Cappuzzo, F., & Rolfo, C. (2015). cMET in NSCLC: Can We Cut off the Head of the Hydra? From the Pathway to the Resistance. Cancers, 7(2), 556-573. https://doi.org/10.3390/cancers7020556