
Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer


Abstract
:1. Introduction
2. Crizotinib

{kind=link}
{kind=link}
| Drugs | Trial | Phase | Prior treatment with ALK-TKI | No. of patients | ORR | PFS | OS | CNS disease | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Crizotinib | PROFILE 1001 | 1 | No | 143 | 60.8% | 9.7 M | estimated 6 M: 87.9% 12 M: 74.8% | [23] | |
| PROFILE 1005 | 2 | No (chemotherapy: Yes) | 439 | 53% | 8.5 M | [24] | |||
| PROFILE 1007 | 3 | No (platinum-based chemotherapy: Yes) | 347 173 vs. 174 | 65% vs. 20% | 7.7 vs. 3.0 M | 12.2 vs. 12.1 M | [25] | ||
| PROFILE 1014 | 3 | No | 343 172 vs. 171 | 74% vs. 45% | 10.9 vs. 7.0 M | probability 12 M: 84% vs. 79% | [26] | ||
| Ceritinib | ASCEND-1 | 1 | Yes (163/246) | 246 | 58% | 8.2 M | 12 M: 65% | ORR: 54% | [27] |
| Alectinib | AF-001JP | 1/2 | No | Phase 1: 24 Phase 2: 46 | 93.5% CR rate: 19.6% | 27.7 M | 12 M: 83% 24 M: 79% | [28,29] | |
| AF-002JG | 1/2 | Yes | Phase 1: 47 | 55% CR rate: 2% | NA | ORR: 52% | [30] | ||
| AP26113 | Gadgeel et al. | 1/2 | Yes | 57 | 72% | 10.9M | 69% improved CNS disease | [31] |
| Mechanisms | Material (patient or cell line) | Number of patients | Agents for overcoming the resistance | Reference | |||
|---|---|---|---|---|---|---|---|
| ALK alteration | ALK amplification | CNG | a patient | 1 of 11 patients (12 samples) | [32] | ||
| ALK amplification + ALK mutation | CNG + G1269A | a patient | 1 of 11 patients (12 samples) | [32] | |||
| CNG + L1196M | H3122CR1 cells (stepwise increase) | TAE684, AP26113, 17-AAG | [33] | ||||
| CNG + 1151Tins | H3122CR2 cells (stepwise increase) | 17-AAG | [34] | ||||
| ALK mutation | L1196M, C1156Y | a patient | 1 | [35] | |||
| Ba/F3 cells (transfected mutation) | |||||||
| F1174L | a patient | 1 | [36] | ||||
| Ba/F3 cells (transfected mutation) | TAE684, 17-AAG | ||||||
| S1206Y, G1202R, L1196M | patients | 3 of 18 patients (19 samples) | [34] | ||||
| S1206Y, G1202R, L1196M, 1151Tins | Ba/F3 cells (transfected mutation) | S1206Y: TAE684, alectinib, 17-AAG; G1202R: TAE684, 17-AAG; L1196M: TAE684, 17-AAG, alectinib; 1151Tins: TAE684, 17-AAG, alectinib (Agents above showing lower IC50 than crizotinib) | [34] | ||||
| L1196M, G1269A | patients | 4 of 11 patients (12 samples) | [32] | ||||
| G1202R | a patient | 1 | [37] | ||||
| L1152R | H3122 cells (transfected mutation) | [38] | |||||
| Bypass track activation | ALK mutation + EGFR activation | L1152R | a patient | 1 | [38] | ||
| secretion of EGFR ligand (amphireglin) | DFCI076 cells (derived from the above-referenced patient) | ALK inhibitor + PF299804 | [38] | ||||
| 1151Tins increased auto-phosphorylation of EGFR | a patient | 1 of 18 patients (19 samples) | [34] | ||||
| EGFR activation | L858R | a patient | 1 | [32] | |||
| retained phosphorylation of EGFR | H3122 cells (external EGF) | crizotinib + PF299804 or gefitinib | [38] | ||||
| H2228 cells, H3122 cells (external EGF, TGF-α and HB-EGF) | [39] | ||||||
| secretion of EGFR ligand (amphiregulin) and ErbB3 ligand (NRG1) | H3122CR3 (stepwise increase) | crizotinib + gefitinib or erlotinib | [34] | ||||
| increased auto-phosphorylation of EGFR | a patient | 1 of 9 patients | [34] | ||||
| Bypass track activation | EGFR + KIT activation | Increased auto-phosphorylation of EGFR KIT amplification + SCF overexperssion | a patient | 1 of 9 patients | [34] | ||
| KIT activation | KIT amplification | a patient | 2 of 18 patients (19 samples) | [34] | |||
| increased phosphorylation of cKIT | H3122 (overexpressed cKIT + external SCF) | crizotinib + imatinib | |||||
| KRAS mutation | G12V | patients | 2 of 11 patients (12 samples) (1 of 2 is intrinsic resistances) | [32] | |||
| CUTO-1 cells (derived from above patient; ALK-, KRAS+) | [32] | ||||||
| IGF-1R activation | increased phosphorylation of IGF-1R | a patient | 1 | [37] | |||
| H3122 (external IGF-1R) | crizotinib + OSI-906 | [37] | |||||

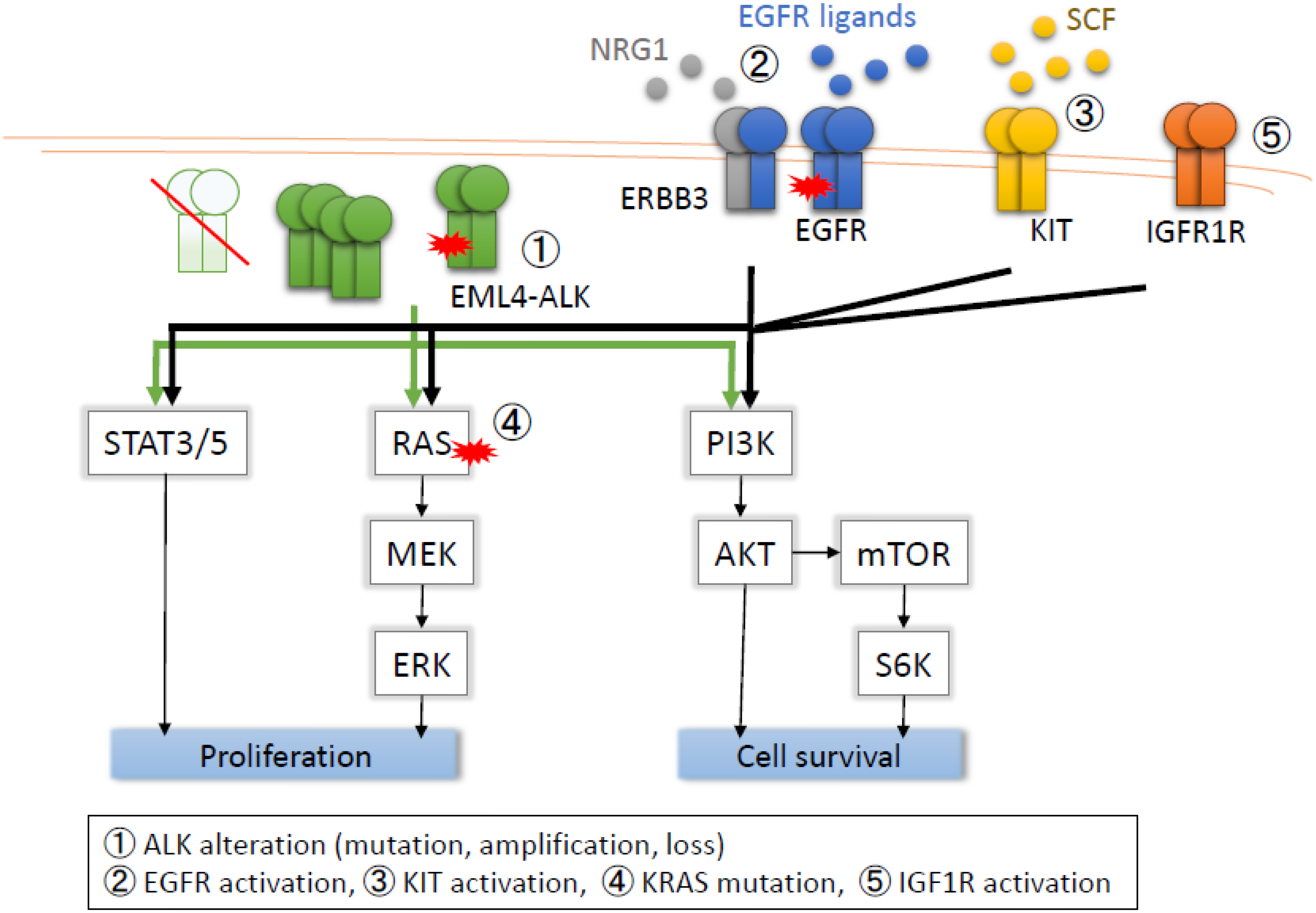
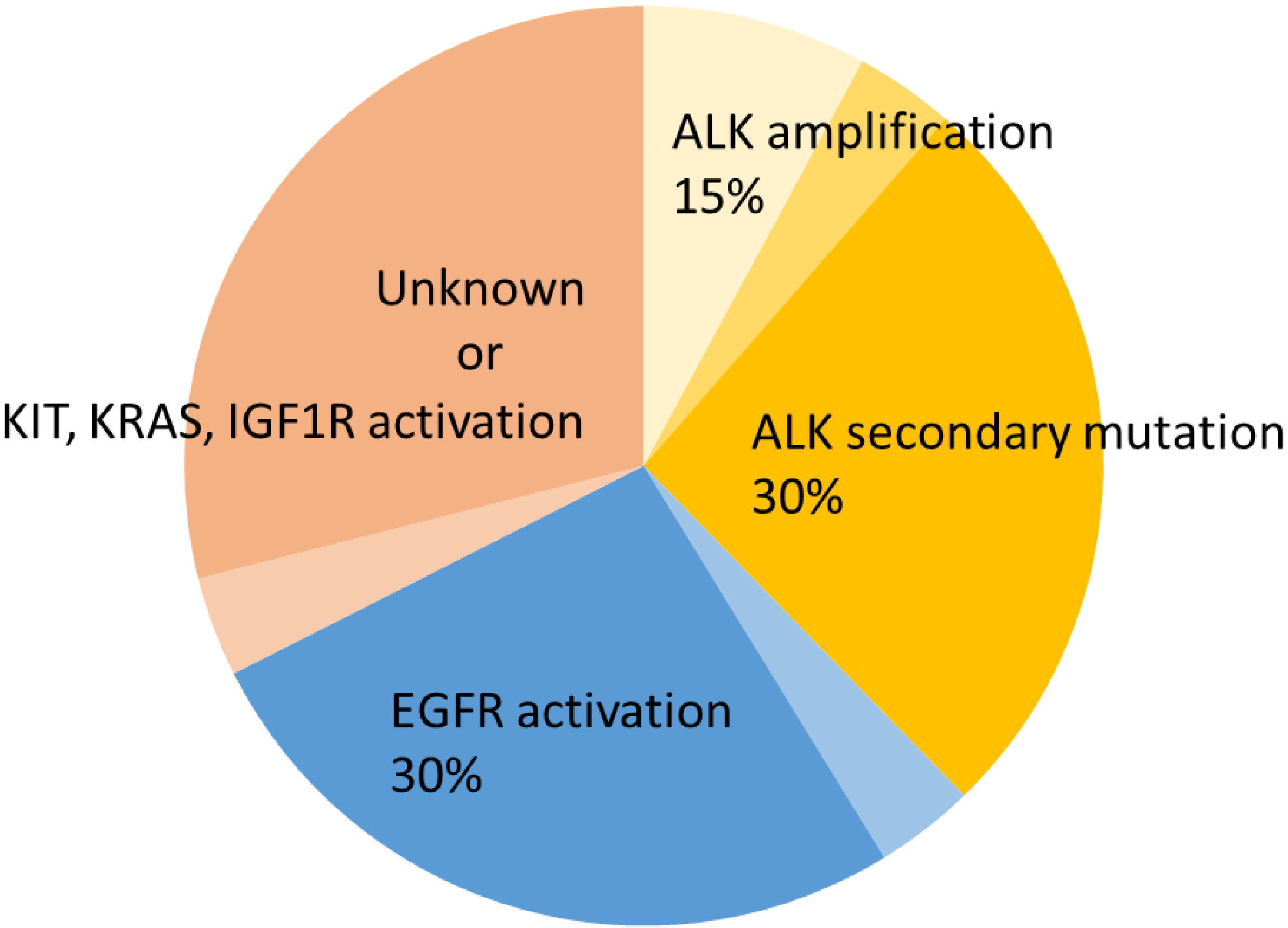
2.1. ALK Secondary Mutations

2.2. ALK Amplification
2.3. Loss of ALK
2.4. EGFR Activation
2.5. cKIT Amplification
2.6. KRAS Mutation
2.7. IGF-1R Activation
3. Ceritinib
4. Alectinib
5. Other Novel ALK Inhibitors
| Drugs | Company | Other activity | Clinical trials | Status |
|---|---|---|---|---|
| Crizotinib (PF-02341066) | Pfizer | MET, ROS1 | Phase 1, 2, 3 | Approved by FDA (Auguet 2011) Clinically available in Japan (March 2012) |
| Ceritinib (LDK378) | Novartis | IGF1R, INSR | Phase 1, 2, 3 | Approved by FDA (May 2014) |
| Alectinib (CH5424802) | Chugai, Roche | RET | Phase 1, 2, 1/2, 3 | Breakthrough Therapy Designation (June 2013) Clinically available in Japan (July 2014) |
| AP26113 | Ariad | EGFR, ROS1 | Phase 1/2 | Breakthrough Therapy Designation (October 2014) |
| ASP3026 | Astellas | ROS1 | Phase 1 | |
| X-376 X-396 | Xcovery | MET | Phase 1 (X-396) | |
| TSR-011 | Tesaro | TRK-A, TRK-B, TRK-C | Phase 1/2a | |
| RXDX-101 | Ignyta | ROS1 TRK-A, TRK-B, TRK-C | Phase 1 | |
| CEP-28122 CEP-37440 | Teva | RSK2, RSK3, RSK4 | Phase 1 (CEP-37440) | |
| PF-06463922 | Pfizer | ROS1 | Phase 1/2 |
6. Conclusions and Future Therapeutic Strategies
Conflicts of Interest
References
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; O’Brien, S.G.; Guilhot, F.; Goldman, J.M.; Hochhaus, A.; Hughes, T.P.; Radich, J.P.; Hatfield, A.K.; Mone, M.; Filian, J.; et al. International randomized study of interferon vs. sti571 (IRIS) 8-year follow up: Sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood 2009, 114. Abstract 1126. [Google Scholar]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- ALK anaplastic lymphoma receptor tyrosine kinase [homo sapiens (human)]. Available online: http://218.245.4.153/cgi-bin/orf/gene.pl?geneid=238&species=9606 (accessed on 20 March 2015).
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Dvorak, C.; Henkle, C.; Ellingham, T.; Perlman, E.J. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999, 59, 2776–2780. [Google Scholar] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Sukov, W.R.; Hodge, J.C.; Lohse, C.M.; Akre, M.K.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C. ALK alterations in adult renal cell carcinoma: Frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod. Pathol. 2012, 25, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Debelenko, L.V.; Raimondi, S.C.; Daw, N.; Shivakumar, B.R.; Huang, D.; Nelson, M.; Bridge, J.A. Renal cell carcinoma with novel vcl-ALK fusion: New representative of ALK-associated tumor spectrum. Mod. Pathol. 2011, 24, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Marino-Enriquez, A.; Ou, W.B.; Weldon, C.B.; Fletcher, J.A.; Perez-Atayde, A.R. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer 2011, 50, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, E.; Togashi, Y.; Kuroda, N.; Sakata, S.; Hatano, S.; Asaka, R.; Yuasa, T.; Yonese, J.; Kitagawa, M.; Mano, H.; et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: Large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer 2012, 118, 4427–4436. [Google Scholar] [CrossRef] [PubMed]
- Jazii, F.R.; Najafi, Z.; Malekzadeh, R.; Conrads, T.P.; Ziaee, A.A.; Abnet, C.; Yazdznbod, M.; Karkhane, A.A.; Salekdeh, G.H. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J. Gastroenterol. 2006, 12, 7104–7112. [Google Scholar] [PubMed]
- Yaakup, H.; Sagap, I.; Fadilah, S.A. Primary oesophageal ki (CD30)-positive ALK+ anaplastic large cell lymphoma of T-cell phenotype. Singapore Med. J. 2008, 49, e289–e292. [Google Scholar] [PubMed]
- Lin, E.; Li, L.; Guan, Y.; Soriano, R.; Rivers, C.S.; Mohan, S.; Pandita, A.; Tang, J.; Modrusan, Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res. 2009, 7, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.; Aigner, A.; Stoica, G.E.; McDonnell, K.; Wellstein, A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J. Biol. Chem. 2002, 277, 14153–14158. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Aigner, A.; Sunitha, I.; Souttou, B.; Malerczyk, C.; Caughey, D.J.; Wen, D.; Karavanov, A.; Riegel, A.T.; et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J. Biol. Chem. 2001, 276, 16772–16779. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Xing, M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011, 71, 4403–4411. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Ahn, M.; Shi, Y.; de Pas, T.M.; Pan-Chyr Yang, P.; Riely, G.J.; Crinò, L.; Evans, T.L.; Liu, X.; Han, J.; et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2012, 30. Abstract 7533. [Google Scholar]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; de Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Kiura, K.; Nishio, M.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yamamoto, N.; Yoshioka, H.; Harada, M.; et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): A single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013, 14, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Seto, T.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yoshioka, H.; Harada, M.; Ohe, Y.; Nogami, N.; et al. Updated data of a phase 1/2 study (AF-001JP) of alectinib, a CNS-penetrant, highly selective ALK inhibitor in ALK-rearranged advanced NSCLC. Radiat. Oncol. 2014. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.M.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant alk-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Takenoyama, M.; Watanabe, S.; Toyozawa, R.; Inamasu, E.; Kojo, M.; Shiraishi, Y.; Morodomi, Y.; Takenaka, T.; Hirai, F.; et al. Dramatic response to crizotinib in an ALK-positive adenocarcinoma patient with disseminated intravascular coagulation. J. Thorac. Oncol. 2013, 8, e96–e98. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamura, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Rudzki, Z.; Walsh, S.; Grigg, A.; Arthur, C.; Taylor, K.; Herrmann, R.; Lynch, K.P.; Hughes, T.P. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002, 99, 3472–3475. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLOS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, K.; Maruvka, Y.E.; Michor, F.; Pao, W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J. Clin. Oncol. 2013, 31, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. Ch5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- IASLC Atlas of ALK Testing in Lung Cancer. Available online: https://www.Iaslc.Org/publications/iaslc-atlas-ALK-testing-lung-cancer (accessed on 20 March 2015).
- Giri, S.; Patel, J.K.; Mahadevan, D. Novel mutations in a patient with ALK-rearranged lung cancer. N. Engl. J. Med. 2014, 371, 1655–1656. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, C.; Wang, S. LDK378: A promising anaplastic lymphoma kinase (ALK) inhibitor. J. Med. Chem. 2013, 56, 5673–5674. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Mehra, R.; Tan, D.S.W.; Felip, E.; Chow, L.Q.M.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; Pas, T.D.; Riely, G.J.; et al. Ceritinib (LDK378) for treatment of patients with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) and brain metastases (BM) in the ascend-1 trial. Neuro-Oncology 2014, 16, v32–v40. [Google Scholar] [CrossRef]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Satoh, Y.; Yoshida, M.; Watanabe, Y.; Kondoh, O.; Sakamoto, H. Alectinib shows potent antitumor activity against ret-rearranged non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Gadgeel, S.; Chiappori, A.; Riely, G.; Lee, R.; Garcia, L.; Tatsuno, M.; Tanaka, T.; Gandhi, L. Safety and efficacy analysis of RO5424802/CH5424802 in anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) patients who have failed crizotinib in a dose-finding phase I study (AF-002JG, NCT01588028). Eur. Cancer Congr. 2013, 49. Abstract 44LBA. [Google Scholar]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Sommers, K.R.; Azada, M.C.; Garon, E.B. Alectinib induces a durable (>15 months) complete response in an ALK-positive non-small cell lung cancer patient who progressed on crizotinib with diffuse leptomeningeal carcinomatosis. Oncologist 2015, 20, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Ajimizu, H.; Kim, Y.H.; Mishima, M. Rapid response of brain metastases to alectinib in a patient with non-small-cell lung cancer resistant to crizotinib. Med. Oncol. 2015. [Google Scholar] [CrossRef]
- Gainor, J.F.; Sherman, C.A.; Willoughby, K.; Logan, J.; Kennedy, E.; Brastianos, P.K.; Chi, A.S.; Shaw, A.T. Alectinib salvages cns relapses in ALK-positive lung cancer patients previously treated with crizotinib and ceritinib. J. Thorac. Oncol. 2015, 10, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Sado, T.; Nishihara, Y.; Fukata, H.; Tajiri, T.; Kita, H. A case of positive for ALK rearrangement lung adenocarcinoma that recurred in meningeal carcinomatosis during administration of crizotinib, improving after switching to alectinib. Ann. Jpn. Respir. Soc. 2014, 4, 139–142. [Google Scholar]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Hirai, F.; Inamasu, E.; Yoshida, T.; Nosaki, K.; Takenaka, T.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Secondary mutations at I1171 in the ALK gene confer resistance to both crizotinib and alectinib. J. Thorac. Oncol. 2014, 9, e86–e87. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, H.; Ichihara, E.; Ohashi, K.; Ochi, N.; Yasugi, M.; Kubo, T.; Minami, D.; Yamane, H.; Sato, A.H.; Kudo, K.; et al. Acquired resistance to a new ALK inhibitor, alectinib in lung cancer. Ann. Oncol. 2014. [Google Scholar] [CrossRef]
- Awad, M.M.; Shaw, A.T. ALK inhibitors in non-small cell lung cancer: Crizotinib and beyond. Clin. Adv. Hematol. Oncol. 2014, 12, 429–439. [Google Scholar] [PubMed]
- Squillace, R.M.; Anjum, R.; Miller, D.; Vodala, S.; Moran, L.; Wang, F.; Clackson, T.; Garner, A.P.; Rivera, V.M. AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. Cancer Res. 2013, 73. Abstract 5655. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.; Salgia, R.; Langer, C.J.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Narasimhan, N.I.; Dorer, D.J.; et al. Updated efficacy and safety of the ALK inhibitor AP26113 in patients (PTS) with advanced malignancies, including ALK+ non-small cell lung cancer (NSCLC). In Proceedings of the 2014 ASCO Annual Meeting, Chicago, IL, USA, 30 May 2014. J. Clin. Oncol. 2014, 32. Abstract 8047. [Google Scholar]
- Patnaik, A.; LoRusso, P.; Ball, H.A.; Bahceci, E.; Yuen, G.; Papadopoulos, K.P.; Kittaneh, M.; Tolcher, A.W. Pharmacokinetics and safety of an oral ALK inhibitor, ASP3026, observed in a phase I dose escalation trial. In Proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013.
- Zou, H.Y.; Engstrom, L.R.; Li, Q.; West Lu, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; Timofeevski, S.; Dinh, D.M.; et al. PF-06463922, a novel brain-penetrating small molecule inhibitor of ALK/ROS1 with potent activity against a broad spectrum of ALK resistant mutations in preclinical models in vitro and in vivo. Mol. Cancer Therapeutics 2013, 12. Abstract C253. [Google Scholar]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Sachdev, J.C.; Infante, J.R.; Mita, M.; Wilcoxen, K.M.; Kansra, V.; Brooks, D.G.; Martell, R.E.; Anthony, S.P. TSR-011, a potent ALK inhibitor with clinical activity in phase I/IIA development. J. Thorac. Oncol. 2013, S8, S618. [Google Scholar]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive ntrk1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Ardini, E.; Menichincheri, M.; de Ponti, C.; Amboldi, N.; Saccardo, M.B.; Texido, G.; Russo, M.; Orsini, P.; Bandiera, T.; Lombardi Borgia, A.; et al. Abstract A243: Characterization of NMS-E628, a small molecule inhibitor of anaplastic lymphoma kinase with antitumor efficacy in ALK-dependent lymphoma and non-small cell lung cancer models. Mol. Cancer Therapeutics 2009. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Saccardo, M.B.; Rusconi, L.; Avanzi, N.; Amboldi, N.; Casero, D.; Cribioli, S.; Isacchi, A.; et al. Abstract A232: In vitro and in vivo activity of NMS-E628 against ALK mutations resistant to Xalkori. Mol. Cancer Therapeutics 2011. [Google Scholar] [CrossRef]
- De Braud, F.G.M.; Pilla, L.; Niger, M.; Damian, S.; Bardazza, B.; Martinetti, A.; Pelosi, G.; Marrapese, G.; Palmeri, L.; Cerea, G.; et al. RXDX-101, an oral pan-TRK, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. Ann. Oncol. 2014, 25, iv148–iv149. [Google Scholar]
- Cheng, M.; Quail, M.R.; Gingrich, D.E.; Ott, G.R.; Lu, L.; Wan, W.; Albom, M.S.; Angeles, T.S.; Aimone, L.D.; Cristofani, F.; et al. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 2012, 11, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isozaki, H.; Takigawa, N.; Kiura, K. Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer. Cancers 2015, 7, 763-783. https://doi.org/10.3390/cancers7020763
Isozaki H, Takigawa N, Kiura K. Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer. Cancers. 2015; 7(2):763-783. https://doi.org/10.3390/cancers7020763
Chicago/Turabian StyleIsozaki, Hideko, Nagio Takigawa, and Katsuyuki Kiura. 2015. "Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer" Cancers 7, no. 2: 763-783. https://doi.org/10.3390/cancers7020763
APA StyleIsozaki, H., Takigawa, N., & Kiura, K. (2015). Mechanisms of Acquired Resistance to ALK Inhibitors and the Rationale for Treating ALK-positive Lung Cancer. Cancers, 7(2), 763-783. https://doi.org/10.3390/cancers7020763