
Role of Parathyroid Hormone-Related Protein Signaling in Chronic Pancreatitis

{kind=link}
{kind=link}
Abstract
:1. Inflammation and Pancreatic Cancer
2. Etiology and Pathology of Chronic Pancreatitis
3. Increased Risk for Pancreatic Cancer in Patients with Chronic Pancreatitis
4. Risk Factors of CP
5. Parathyroid Hormone-Related Protein Biology
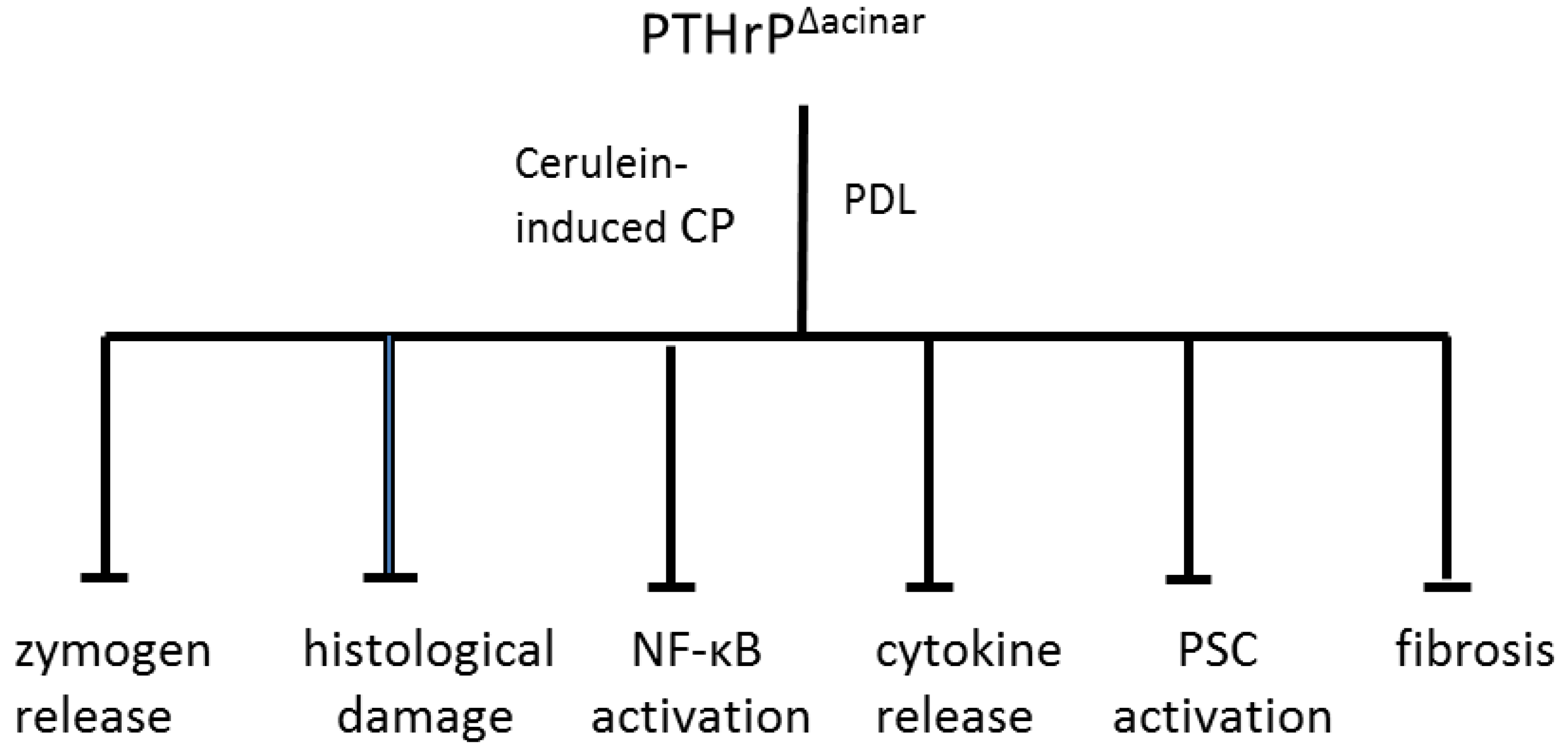
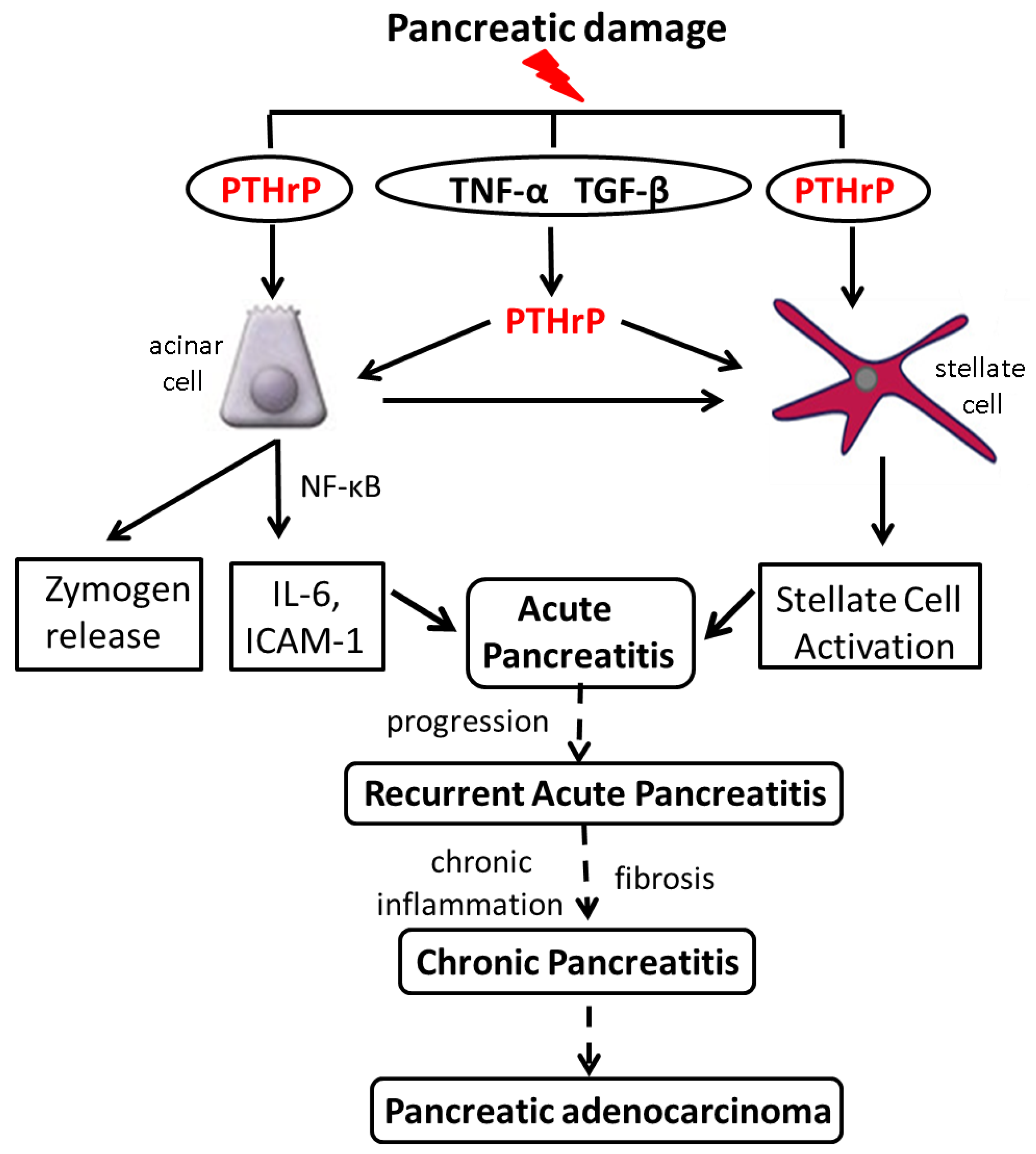
6. Increased PTHrP Levels after Acinar Cell and PSC Injury Lead to Elevated Cytokine and ECM Protein Levels
7. PTHrP May Play a Role in Sensitizing Pancreatic Cells to the Effects of Alcohol
8. PTHrP Levels Are Increased in Mouse Models of AP and CP
9. PTHrP May Function as a CP Modifier Molecule

10. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cancer Facts and Figures 2014, American Cancer Society. Available online: http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf (accessed on 10 June 2015).
- Whitcomb, D.C. Inflammation and Cancer V. Chronic pancreatitis and pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G315–G319. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Neoptolemos, J.P. Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell 2012, 148, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Hruban, R.H.; Klein, A.P. Familial pancreatic cancer. Arch. Pathol. Lab. Med. 2009, 133, 365–374. [Google Scholar] [PubMed]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Bracci, P.M. Obesity and pancreatic cancer: Overview of epidemiologic evidence and biologic mechanisms. Mol. Carcinog. 2012, 51, 53–63. [Google Scholar] [CrossRef] [PubMed]
- What are the Risk Factors for Pancreatic Cancer? American Cancer Society. Available online: http://www.cancer.org/cancer/pancreaticcancer/detailedguide/pancreatic-cancer-risk-factors (accessed on 10 June 2015).
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a key event in cancer development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Inflammation and pancreatic cancer: Disease promoter and new therapeutic target. J. Gastroenterol. 2014, 49, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Etemad, B.; Whitcomb, D.C. Chronic pancreatitis: Diagnosis, classification, and new genetic developments. Gastroenterology 2001, 120, 682–707. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Whitcomb, D.C. Hereditary pancreatitis: A model for inflammatory diseases of the pancreas. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Bridle, K.R.; Wells, L.D.; Marcu, M.; Ramm, G.A. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig. Dis. Sci. 2000, 45, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.; Conwell, D.L.; Zuccaro, G. Pathogenesis of chronic pancreatitis: An evidence-based review of past theories and recent developments. Am. J. Gastroenterol. 2004, 99, 2256–2270. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Whitcomb, D.C. The role of alcohol and smoking in pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Spanier, B.W.M.; Dijkgraaf, M.G.W.; Bruno, M.J. Epidemiology, aetiology and outcome of acute and chronic pancreatitis: An update. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Apte, M.V.; Keim, V.; Wilson, J.S. Chronic pancreatitis: Challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007, 132, 1557–1573. [Google Scholar] [CrossRef] [PubMed]
- Forsmark, C.E. Management of chronic pancreatitis. Gastroenterology 2013, 144, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Trikudanathan, G.; Navaneethan, U.; Vege, S.S. Modern treatment of patients with chronic pancreatitis. Gastroenterol. Clin. N. Am. 2012, 41, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, M.; Tashiro, M. 4. Chronic pancreatitis and pancreatic cancer, lifestyle-related diseases. Intern. Med. 2007, 46, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren, S.A.; Domellof, L. Pancreatitis and the risk of pancreatic . N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.; Maisonneuve, P.; DiMagno, E.; Elitsur, Y.; Gates, L.; Perrault, J.; Whitcomb, D. Hereditary pancreatitis and the risk of pancreatic cancer. J. Natl. Cancer Inst. 1997, 89, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C.; Pogue-Geile, K. Pancreatitis as a risk for pancreatic cancer. Gastroenterol. Clin. N. Am. 2002, 31, 663–678. [Google Scholar] [CrossRef]
- Rossi, L.; Pfützer, R.L.; Parvin, S.; Ali, L.; Sattar, S.; Kahn, A.K.; Gyr, K.; Whitcomb, D.C. SPINK1/PSTI mutations are associated with tropical pancreatitis in Bangladesh: A preliminary report. Pancreatology 2001, 1, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C.; LaRusch, J.; Krasinska, A.M.; Yu, L. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat. Genet. 2012, 44, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Dufour, M.C.; Adamson, M.D. The epidemiology of alcohol-induced pancreatitis. Pancreas 2003, 27, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Pitchumoni, C.S.; Glasser, M.; Saran, R.M.; Panchacharam, P.; Thelmo, W. Pancreatic fibrosis in chronic alcoholics and nonalcoholics without clinical pancreatitis. Am. J. Gastroenterol. 1984, 79, 382–388. [Google Scholar] [PubMed]
- Sand, J.; Lankisch, P.G.; Nordback, I. Alcohol consumption in patients with acute or chronic pancreatitis. Pancreatology 2007, 7, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.J.; Gorelick, F.S.; Gerloff, A.; Lugea, A. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig. Dis. 2010, 28, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B.; Mullhaupt, B.; Cavallini, G.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L.; Frulloni, L.; et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 2005, 54, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Tamakoshi, A.; Hayakawa, T.; Ogawa, M.; Ohno, Y. Cigarette smoking as a risk factor for chronic pancreatitis: A case-control study in Japan. Research Committee on intractable Pancreatic Diseases. Pancreas 2000, 21, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Hawes, R.H.; Brand, R.E.; Anderson, M.A.; Money, M.E.; Banks, P.A.; Bishop, M.D.; Baillie, J.; Sherman, S.; DiSario, J.; et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch. Intern. Med. 2009, 169, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Levy, P.; Mathurin, P.; Roqueplo, A.; Rueff, B.; Bernades, P. A multidimensional case-control study of dietary, alcohol, and tobacco habits in alcoholic men with chronic pancreatitis. Pancreas 1995, 10, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Noel-Jorand, M.C.; Bras, J. A comparison of nutritional profiles of patients with alcohol related pancreatitis and cirrhosis. Alcohol. Alcoholism. 1994, 29, 65–74. [Google Scholar] [PubMed]
- Reed, A.; Gorelick, F. Animal Models of Chronic Pancreatitis. Pancreapedia: Exocrine Pancreas Knowl. Base 2014. [Google Scholar] [CrossRef]
- Strewler, G.I. Mechanisms of disease: The physiology of parathyroid hormone-related protein. N. Engl. J. Med. 2000, 342, 177–185. [Google Scholar] [PubMed]
- Wysolmerski, J.J. Parathyroid hormone-related protein: An update. Clin. Endocrinol. Metab. 2012, 97, 2947–2956. [Google Scholar] [CrossRef] [PubMed]
- Mannstadt, M.; Jüppner, H.; Gardella, T.J. Receptors for PTH and PTHrP. Am. J. Physiol. 1999, 277, F665–F675. [Google Scholar] [PubMed]
- Henderson, J.E.; Amikuza, N.; Warshawsky, H.; Biasotto, D.; Lanske, B.M.; Goltzman, D.; Karaplis, A.C. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol. Cell. Biol. 1995, 15, 4064–4075. [Google Scholar] [PubMed]
- Massfelder, T.; Dann, P.; Wu, T.L.; Vasavada, R.; Helwig, J.-J.; Stewart, A.F. Opposing mitogenic and antimitogenic actions of parathyroid hormone-related protein in vascular smooth muscle cells: A critical role for nuclear targeting. Proc. Natl. Acad. Sci. USA 1997, 94, 13630–13635. [Google Scholar] [CrossRef] [PubMed]
- Falzon, M.; Du, P. Enhanced growth of MCF-7 breast cancer cells overexpressing parathyroid hormone-related protein. Endocrinology 2000, 141, 1882–1892. [Google Scholar] [PubMed]
- Bhatia, V.; Mula, R.V.; Weigel, N.L.; Falzon, M. Parathyroid hormone-related protein regulates cell survival pathways via integrin alpha6beta4-mediated activation of phosphatidylinositol 3-kinase/Akt signaling. Mol. Cancer Res. 2009, 7, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Mula, R.V.; Bhatia, V.; Falzon, M. PTHrP promotes colon cancer cell migration and invasion in an integrin α6β4-dependent manner through activation of Rac1. Cancer Lett. 2010, 298, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Rizk-Rabin, M.; Assie, G.; Rene-Corail, F.; Perlemoine, K.; Hamzaoui, H.; Tissier, F.; Lieberherr, M.; Bertagna, X.; Bertherat, J.; Bouizar, Z. Differential expression of parathyroid hormone-related protein in adrenocortical tumors: Autocrine/paracrine effects on the growth and signaling pathways in H295R cells. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Vasavada, R.C.; Cavaliere, C.; D’Ercole, A.J.; Dann, P.; Burtis, W.J.; Madlener, A.L.; Zawalich, K.; Zawalich, W.; Philbrick, W.; Stewart, A.F. Overexpression of parathyroid hormone-related protein in the pancreatic islet of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. J. Biol. Chem. 1996, 271, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Cebrian, A.; Garcia-Ocano, A.; Takane, K.K.; Sipula, D.; Stewart, A.F.; Vasavada, R.C. Overexpression of parathyroid hormone-related protein inhibits pancreatic β-cell death in vivo and in vitro. Diabetes 2002, 51, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Mozar, A.; Kondegowda, N.G.; Pollack, I.; Fenutria, R.; Vasavada, R.C. The role of PTHrP in pancreatic beta-cells and implications for diabetes pathophysiology and treatment. Clin. Rev. Bone Miner. Metab. 2014, 12, 165–177. [Google Scholar] [CrossRef]
- Kim, H. Cerulein pancreatitis: Oxidative stress, inflammation, and apoptosis. Gut Liver 2008, 2, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and Alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Siech, M.; Zhou, Z.; Zhou, S.; Bair, B.; Alt, A.; Hamm, S.; Gross, H.; Mayer, J.; Beger, H.G.; Tian, X.; et al. Stimulation of stellate cells by injured acinar cells: A model of acute pancreatitis induced by alcohol and fat (VLDL). Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1163–G1171. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Carangelo, R.; Miller, L.R.; Gorelick, F. Effect of ethanol on cholecystokinin-stimulated zymogen conversion in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 1996, 270, G171–G175. [Google Scholar]
- Apte, M.V.; Wilson, J.S. Stellate cell activation in alcoholic pancreatitis. Pancreas 2003, 27, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Kim, S.O.K.; Aronson, J.F.; Chao, C.; Hellmich, M.R.; Falzon, M. Role of parathyroid hormone-related protein in the pro-inflammatory and pro-fibrogenic response associated with acute pancreatitis. Reg. Pept. 2012, 175, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Ortego, M.; Esbrit, P.; Hernández-Presa, M.A.; Ortega, L.; Egido, J. Possible role of parathyroid hormone-related protein as a proinflammatory cytokine in atherosclerosis. Stroke 2003, 34, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Rámila, D.; Ardura, J.A.; Esteban, V.; Ortega, A.; Ruiz-Ortega, M.; Bosch, R.J.; Esbrit, P. Parathyroid hormone-related protein promotes inflammation in the kidney with an obstructed ureter. Kidney Int. 2008, 873, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sakamotoa, H.; Horiuchi, T.; Yamamoto, S.; Suematsu, A.; Oda, H.; Koshihara, Y. Involvement of prostaglandin E2 in interleukin-1α-induced parathyroid hormone-related peptide production in synovial fibroblasts of patients with rheumatoid arthritis. J. Clin. Endocrinol. Metab. 2001, 86, 3272–3278. [Google Scholar] [PubMed]
- Omary, M.B.; Lugea, A.L.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic disease. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Rastellini, C.; Han, S.; Aronson, J.F.; Greeley, G.H.; Falzon, M. Acinar cell-specific knockout of the PTHrP gene decreases the pro-inflammatory and pro-fibrotic response in pancreatitis. Am. J. Physiol. Gastrointest. Physiol. 2014, 307, G533–G549. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.A.; Kuchmiy, A.; Grivennikov, S.I.; Tumanov, A.V.; Kuprash, D.V.; Nedospasov, S.A. Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: Mouse models. Cytokine Growth Factor Rev. 2008, 19, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.G.; Fink, G.S.; Franz, M.G. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch. Surg. 1995, 130, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Grewal, H.; Kotb, M.; Din, A.M.E.; Ohman, M.; Salem, A.; Gaber, L.; Gaber, A. Induction of tumor necrosis factor in severe acute pancreatitis and its subsequent reduction after hepatic passage. Surgery 1994, 115, 213–221. [Google Scholar] [PubMed]
- Hughes, C.B.; Grewall, H.P.; Gaber, L.W.; Kotb, M.; El-Din, A.B.; Mann, L.; Gaber, A.O. Anti-TNFα therapy improves survival and ameliorates the pathophysiologic sequelae in acute pancreatitis in the rat. Am. J. Surg. 1996, 171, 274–280. [Google Scholar] [CrossRef]
- Gu, H.; Werner, J.; Bergmann, F.; Whitcomb, D.C.; Büchler, M.W.; Fortunato, F. Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Gukovsky, I.; Zaninovic, V.; Song, M.; Sandoval, D.; Gukovsky, S.; Pandol, S.J. Pancreatic acinar cells produce, release, and respond to Tumor Necrosis Factor-α. Role in regulating cell death and pancreatitis. J. Clin. Investig. 1997, 100, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Treede, I.; Braun, A.; Jeliaskova, P.; Giese, T.; Füllekrug, J.; Griffiths, G.; Stremmel, W.; Ehehalt, R. TNF-α-induced up-regulation of pro-inflammatory cytokines is reduced by phosphatidylcholine in intestinal epithelial cells. BMC Gastroenterol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Falzon, M. The University of Texas Medical Branch: Galveston, TX, USA, Unpublished work. 2015.
- Martin, T.J.; Moseley, J.M.; Williams, E.D. Parathyroid hormone-related protein: Hormone and cytokine. J. Endocrinol. 1997, 154, S23–S37. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Fine, D.R. Fibrogenesis in the pancreas after acinar cell injury. Scand. J. Surg. 2005, 94, 108–111. [Google Scholar] [PubMed]
- Yu, J.H.; Kim, K.H.; Kim, H. SOCS 3 and PPAR-γ ligands inhibit the expression of IL-6 and TGF-β1 by regulating JAK2/STAT3 signaling in pancreas. Int. J. Biochem. Cell. Biol. 2008, 40, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cao, Y.; Yang, W.; Duan, C.; Aronson, J.F.; Rastellini, C.; Chao, C.; Hellmich, M.R.; Ko, T.C. BMP2 inhibits TGF-β-induced pancreatic stellate cell activation and extracellular matrix formation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G804–G813. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic stellate cells respond to inflammatory cytokines: Potential role in chronic pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Nagashio, Y.; Ueno, H.; Imamura, M.; Asaumi, H.; Watanabe, S.; Yamaguchi, T.; Taguchi, M.; Tashiro, M.; Otsuki, M. Inhibition of transforming growth factor β decreases pancreatic fibrosis and protects the pancreas against chronic injury in mice. Lab. Investig. 2004, 84, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 60, 1787–1798. [Google Scholar] [CrossRef]
- Vogelmann, R.; Ruf, D.; Wagner, M.; Adler, G.; Menke, A. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-beta1 transgenic mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G164–G172. [Google Scholar] [PubMed]
- Yoo, B.M.; Yeo, M.; Oh, T.Y.; Choi, J.H.; Kim, W.W.; Kim, J.H.; Cho, S.W.; Kim, S.J.; Hahm, K.B. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas 2005, 30, e71–e79. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.D.; Williamson, R.C.; Foster, C.S. Expression of transforming growth factor-beta 1 in chronic pancreatitis. Digestion 1995, 56, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, J.L.; Deviere, J.; Resibois, A.; Rickaert, F.; Vertongen, P.; Ohtani, H.; Cremer, M.; Miyazono, K.; Robberecht, P. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology 1995, 108, 1873–1881. [Google Scholar] [CrossRef]
- Friess, H.; Lu, Z.; Riesle, E.; Uhl, W.; Bründler, A.M.; Horvath, L.; Gold, L.I.; Korc, M.; Büchler, M.W. Enhanced expression of TGF-βs and their receptors in human acute pancreatitis. Ann. Surg. 1998, 227, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Karaplis, A.; Sohn, P. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor β (TGF-β) on endochondral bone formation. J. Cell. Biol. 1999, 145, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, W.; Gao, X.; Zhang, G.; Falzon, M.; Townsend, C.M.; Hellmich, M.R.; Ko, T.C. PTHrP is a novel mediator for TGF-β-induced apoptosis. Reg. Pept. 2013, 184, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, Y.; Daniluk, J.; Gaiser, S.; Chu, J.; Wang, H.; Li, Z.S.; Logsdon, C.D.; Ji, B. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 2013, 144, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Gukovsky, I.; Gukovskaya, A. Nuclear factor-kappaB in pancreatitis: Jack-of-all-trades, but which one is more important? Gastroenterology 2013, 144, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Altavilla, D.; Famulari, C.; Passaniti, M.; Galeano, M.; Macri, A.; Seminara, P.; Minutoli, L.; Marini, H.; Calo, M.; Venuti, F.S.; et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab. Investig. 2003, 83, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Rakonczay, Z.; Hegyi, P.; Takacs, T.; McCarroll, J.; Saluja, A.K. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut 2008, 57, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Steinle, A.U.; Weidenbach, H.; Wagner, M.; Adler, G.; Schmid, R.M. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999, 116, 420–430. [Google Scholar] [CrossRef]
- Vaquero, E.; Gukovsky, I.; Zaninovic, V.; Gukovskaya, A.S.; Pandol, S.J. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1197–G1208. [Google Scholar] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Grady, T.; Liang, P.; Ernst, S.A.; Logsdon, C.D. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 1997, 113, 1966–1975. [Google Scholar] [CrossRef]
- Ji, B.; Bi, Y.; Simeone, D.; Mortensen, R.M.; Logsdon, C.D. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 2001, 121, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.S.; Ruggeri, B.; Barber, M.T.; Biswas, S.; Miknyocki, S.; Waldman, S.A. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J. Clin. Investig. 1997, 100, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.A.; Criddle, D.N.; Sherwood, M.; Chvanov, M.; Mukherjee, R.; McLaughlin, E.; Booth, D.; Gerasimenko, J.V.; Raraty, M.G.; Ghaneh, P.; et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 2008, 135, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Adler, G. Regulation of human pancreatic secretion. Digestion 1997, 58, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Owyang, C. Physiological mechanisms of cholecystokinin action on pancreatic secretion. Am. J. Physiol. 1996, 271, G1–G7. [Google Scholar] [PubMed]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R.; et al. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Karne, S.; Kolodecik, T.; Gorelick, F.S. Alcohols enhance caerulein-induced zymogen activation in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G501–G507. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.J.; Periskic, S.; Gukovsky, I.; Zaninovic, V.; Jung, Y.; Zong, Y.; Solomon, T.E.; Gukovskaya, A.S.; Tsukamoto, H. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 1999, 117, 706–716. [Google Scholar] [CrossRef]
- Aghdassi, A.A.; Mayerle, J.; Christochowitz, S.; Weiss, F.U.; Sendler, M.; Lerch, M.M. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of acute and chronic pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.; Pirola, R.; Wilson, J. The fibrosis of chronic pancreatitis: New insights into the role of pancreatic stellate cells. Antioxid. Redox Signal. 2011, 15, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Braganza, J.M.; Lee, S.H.; McCloy, R.F.; McMahon, M.J. Chronic pancreatitis. Lancet 2011, 377, 1184–1197. [Google Scholar] [CrossRef]
- Kloppel, G.; Detlefsen, S.; Feyerabend, B. Fibrosis of the pancreas: The initial tissue damage and the resulting pattern. Virchows Arch. 2004, 445, 1–8. [Google Scholar] [PubMed]
- He, B.; Deckelbaum, R.A.; Miao, D.; Lipman, M.L.; Pollack, M.; Goltzman, D.; Karaplis, A.C. Tissue-specific targeting of the PTHrP gene: The generation of mice with floxed alleles. Endocrinology 2001, 142, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Karaplis, A.C.; Huang, D.C.; Siegel, P.M.; Camirand, A.; Yang, X.F.; Muller, W.J.; Kremer, R. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 2011, 121, 4655–4669. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; He, B.; Jiang, Y.; Kobayashi, T.; Sorocéanu, M.A.; Zhao, J.; Su, H.; Tong, X.; Amizuka, N.; Gupta, A.; et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J. Clin. Investig. 2005, 115, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.M.; Oliver-Krasinski, J.; de Leon, D.D.; Farzad, C.; Hong, N.; Leach, S.D.; Stoffers, D.A. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet β cell, regeneration. J. Clin. Investig. 2007, 117, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Contet, C. Gene expression under the influence: Transcriptional profiling of ethanol in the brain. Curr. Psychopharmacol. 2012, 1, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Yuan, Y.; Liu, J.; Chen, G. Pioglitazone, a PPAR-γ activator, attenuates the severity of cerulein-induced acute pancreatitis by modulating early growth response-1 transcription factor. Trans. Res. 2012, 160, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Luparello, C. Parathyroid hormone-related protein (PTHrP): A key regulator of life/death decisions by tumor cells with potential clinical applications. Cancers 2011, 3, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Camirand, A.; Fadhil, I.; Luco, A.-E.; Ochietti, B.; Kremer, R.B. Enhancement of taxol, doxorubicin and zoledronate anti-proliferation action on triple-negative breast cancer cells by a PTHrP blocking monoclonal antibody. Am. J. Cancer Res. 2013, 3, 500–508. [Google Scholar] [PubMed]
- Dackiw, A.; Pan, J.; Xu, G.; Yeung, S.-C.J. Modulation of parathyroid hormone-related protein levels (PTHrP) in anaplastic thyroid cancer. Surgery 2005, 138, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Gallwitz, W.E.; Guise, T.A.; Mundy, G.R. Guanosine nucleotides inhibit different syndromes of PTHrP excess caused by human cancers in vivo. J. Clin. Investig. 2002, 110, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falzon, M.; Bhatia, V. Role of Parathyroid Hormone-Related Protein Signaling in Chronic Pancreatitis. Cancers 2015, 7, 1091-1108. https://doi.org/10.3390/cancers7020826
Falzon M, Bhatia V. Role of Parathyroid Hormone-Related Protein Signaling in Chronic Pancreatitis. Cancers. 2015; 7(2):1091-1108. https://doi.org/10.3390/cancers7020826
Chicago/Turabian StyleFalzon, Miriam, and Vandanajay Bhatia. 2015. "Role of Parathyroid Hormone-Related Protein Signaling in Chronic Pancreatitis" Cancers 7, no. 2: 1091-1108. https://doi.org/10.3390/cancers7020826
APA StyleFalzon, M., & Bhatia, V. (2015). Role of Parathyroid Hormone-Related Protein Signaling in Chronic Pancreatitis. Cancers, 7(2), 1091-1108. https://doi.org/10.3390/cancers7020826