
Oncogenic BRAF(V600E) Induces Clastogenesis and UVB Hypersensitivity

Abstract
:1. Introduction
2. Results and Discussion
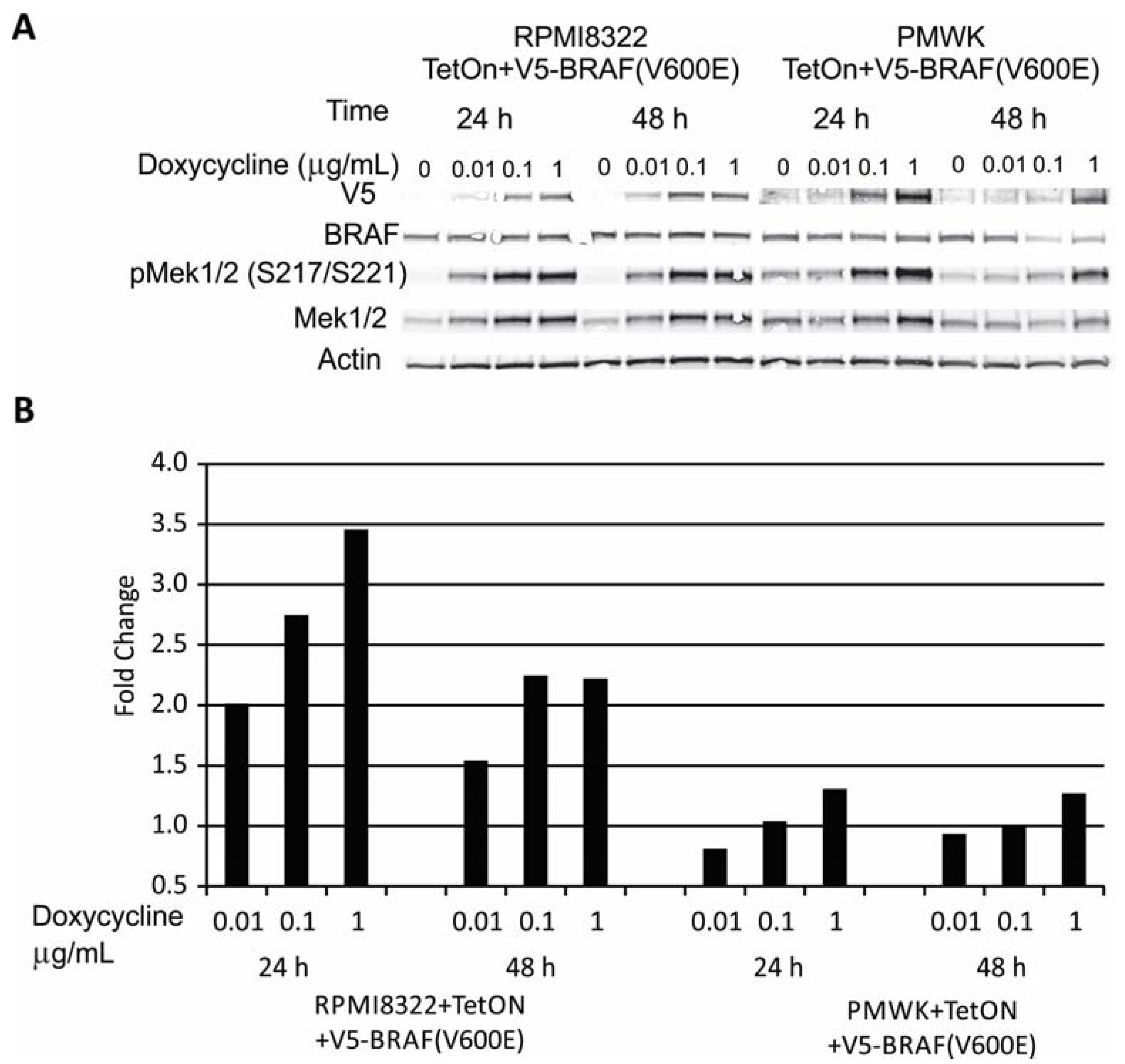
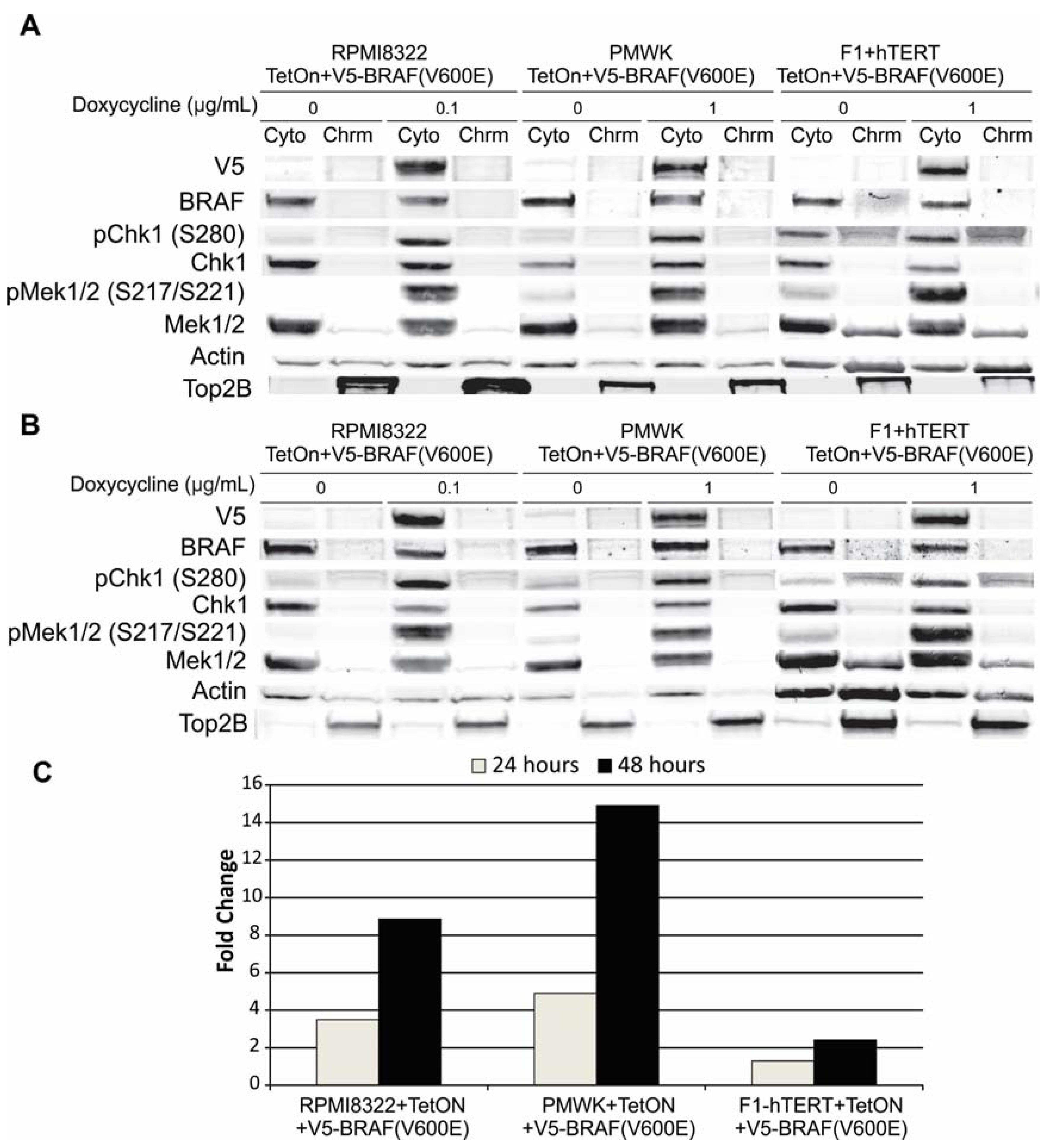
2.1. Engineering and Validation of Cell Lines


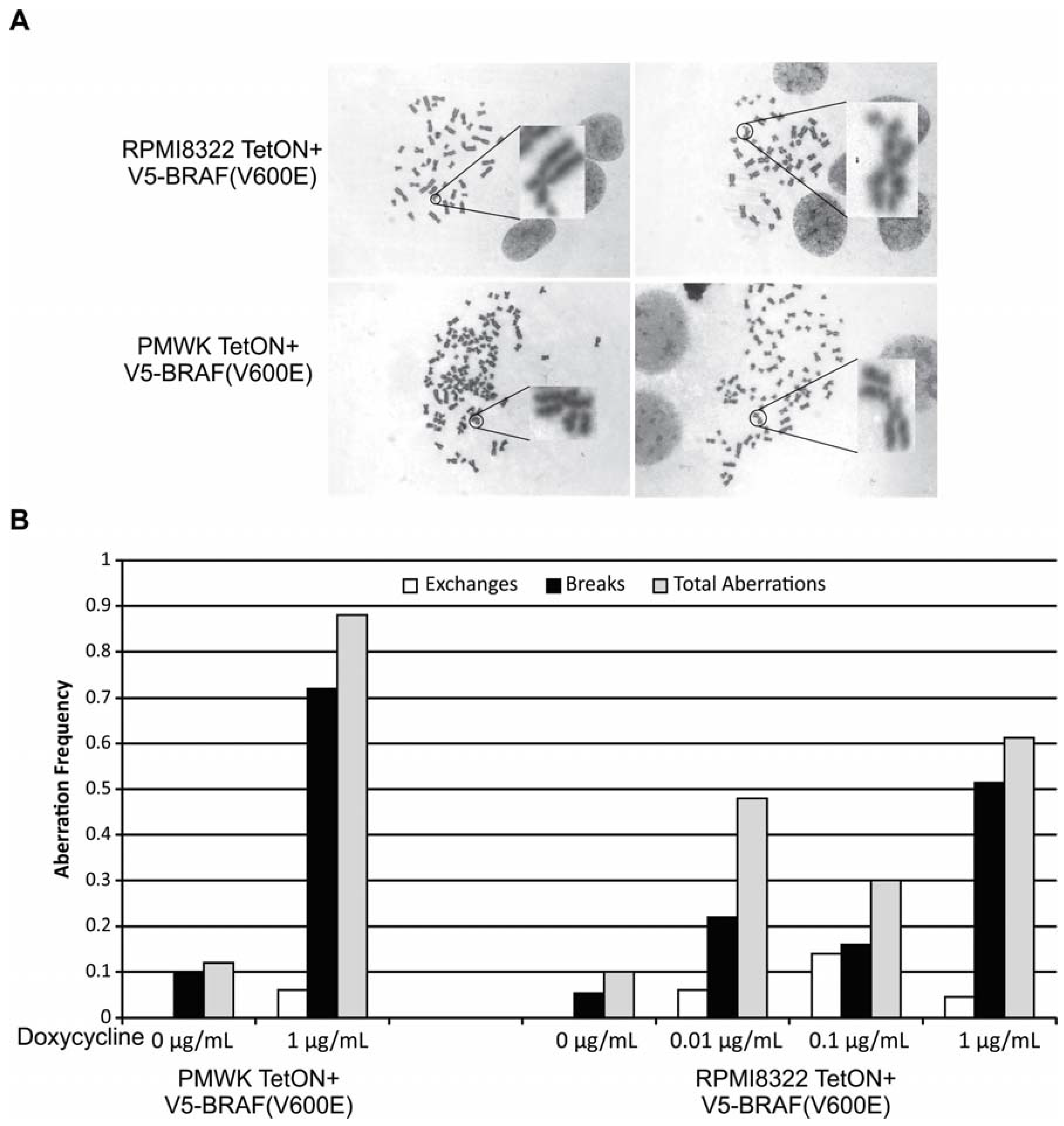
2.2. Oncogenic BRAF Induced Chromosomal Aberrations

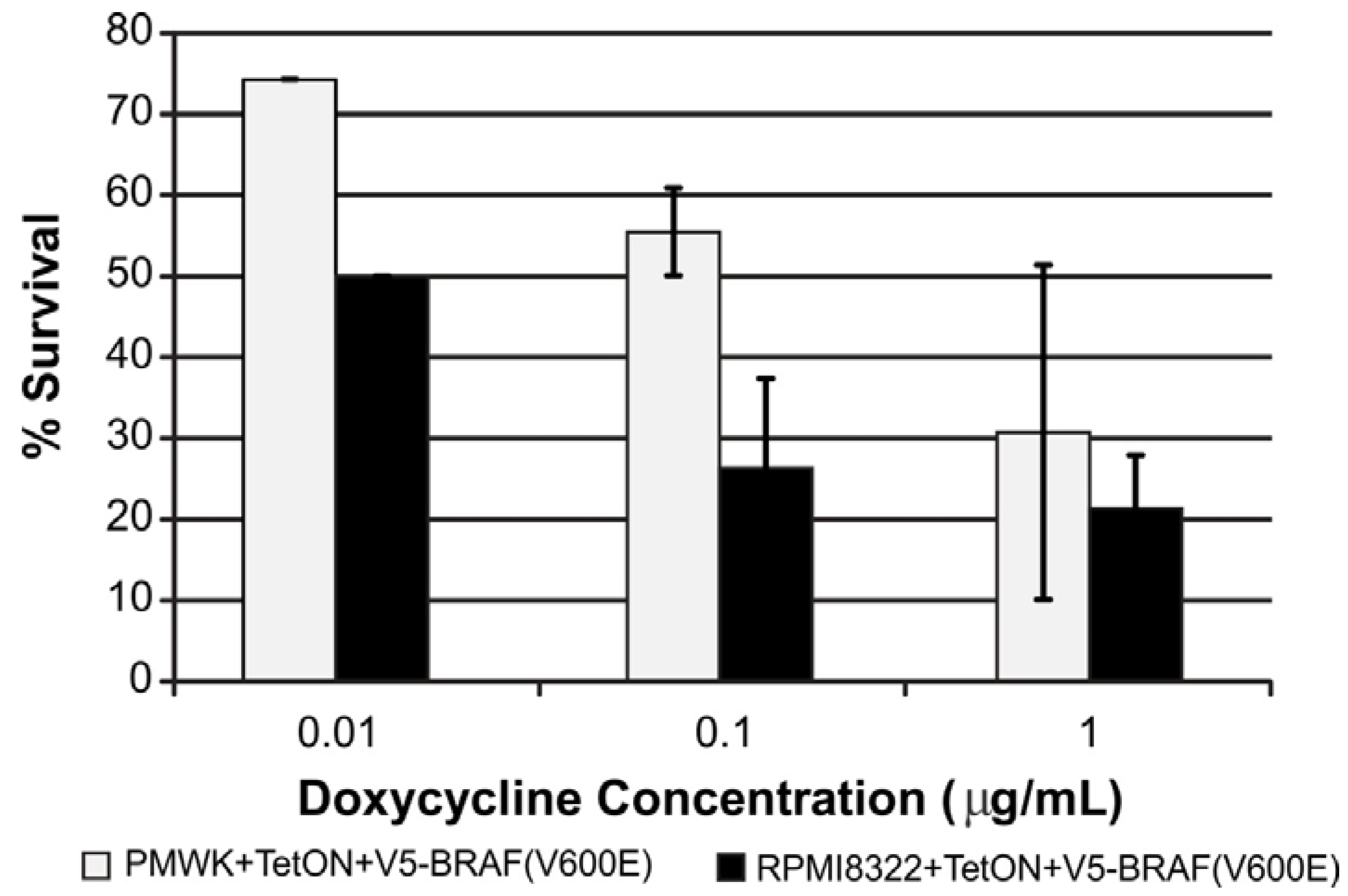
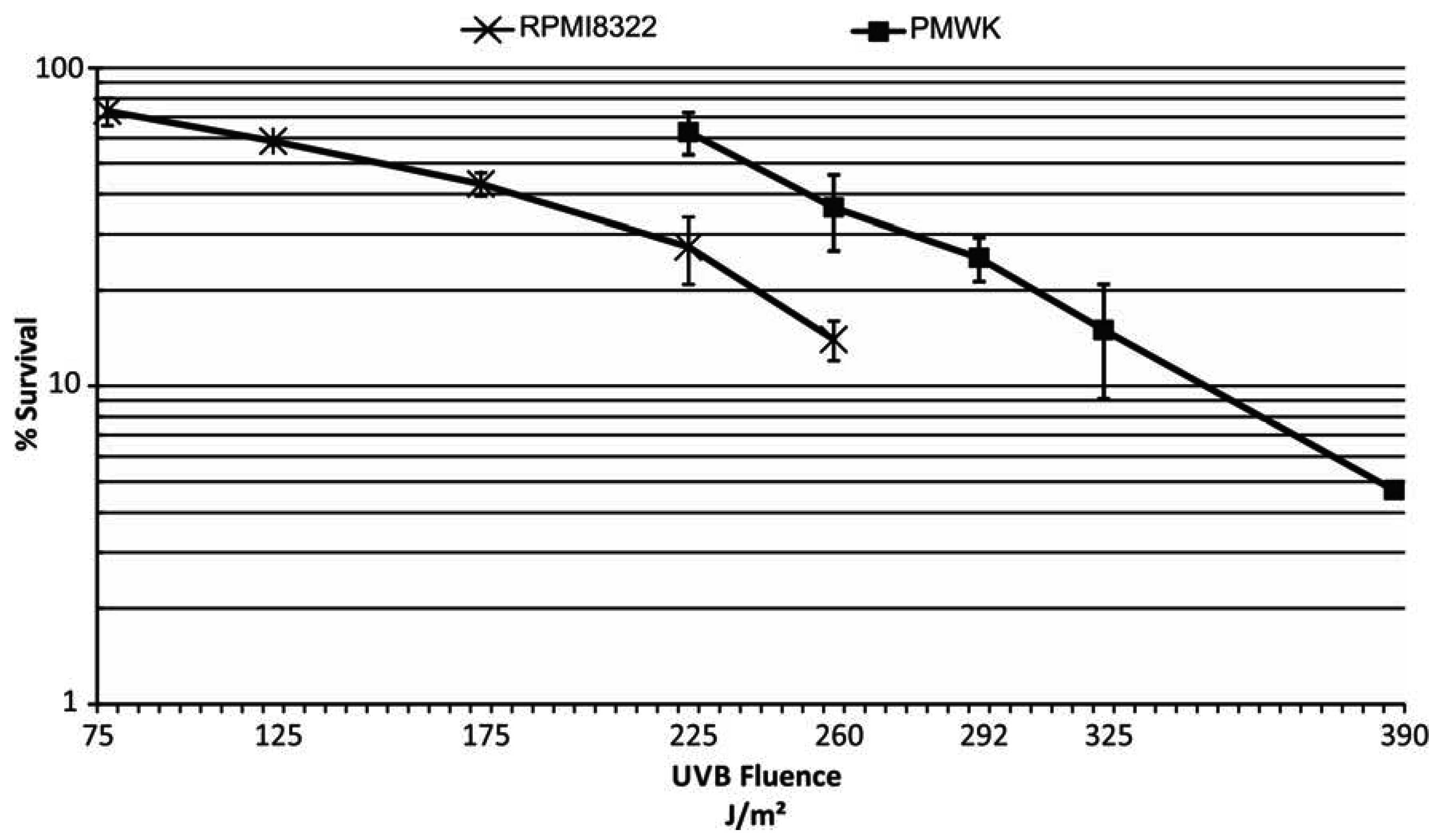
2.3. UVB Sensitivity of Cell Lines

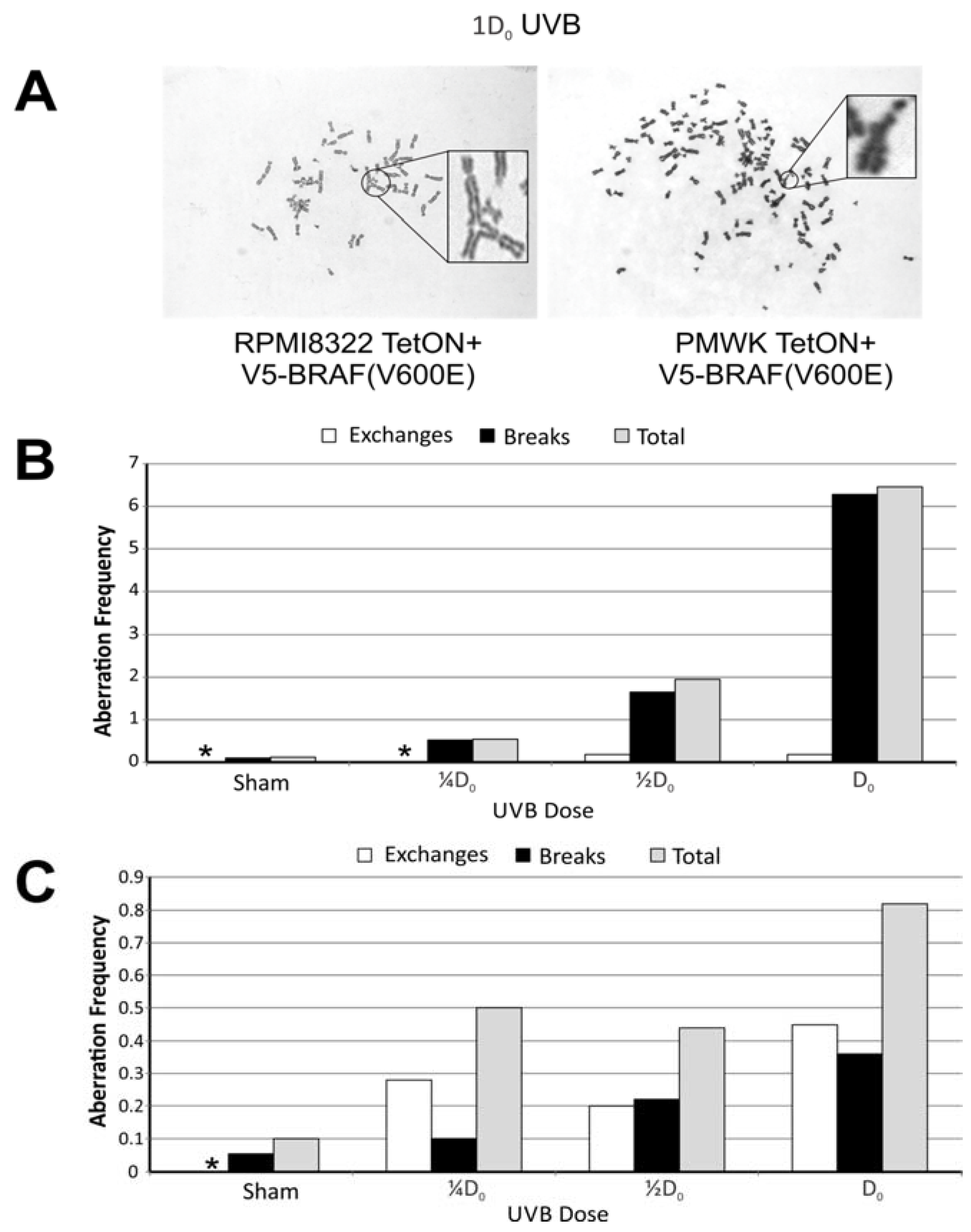
2.4. UVB Induced Aberrations

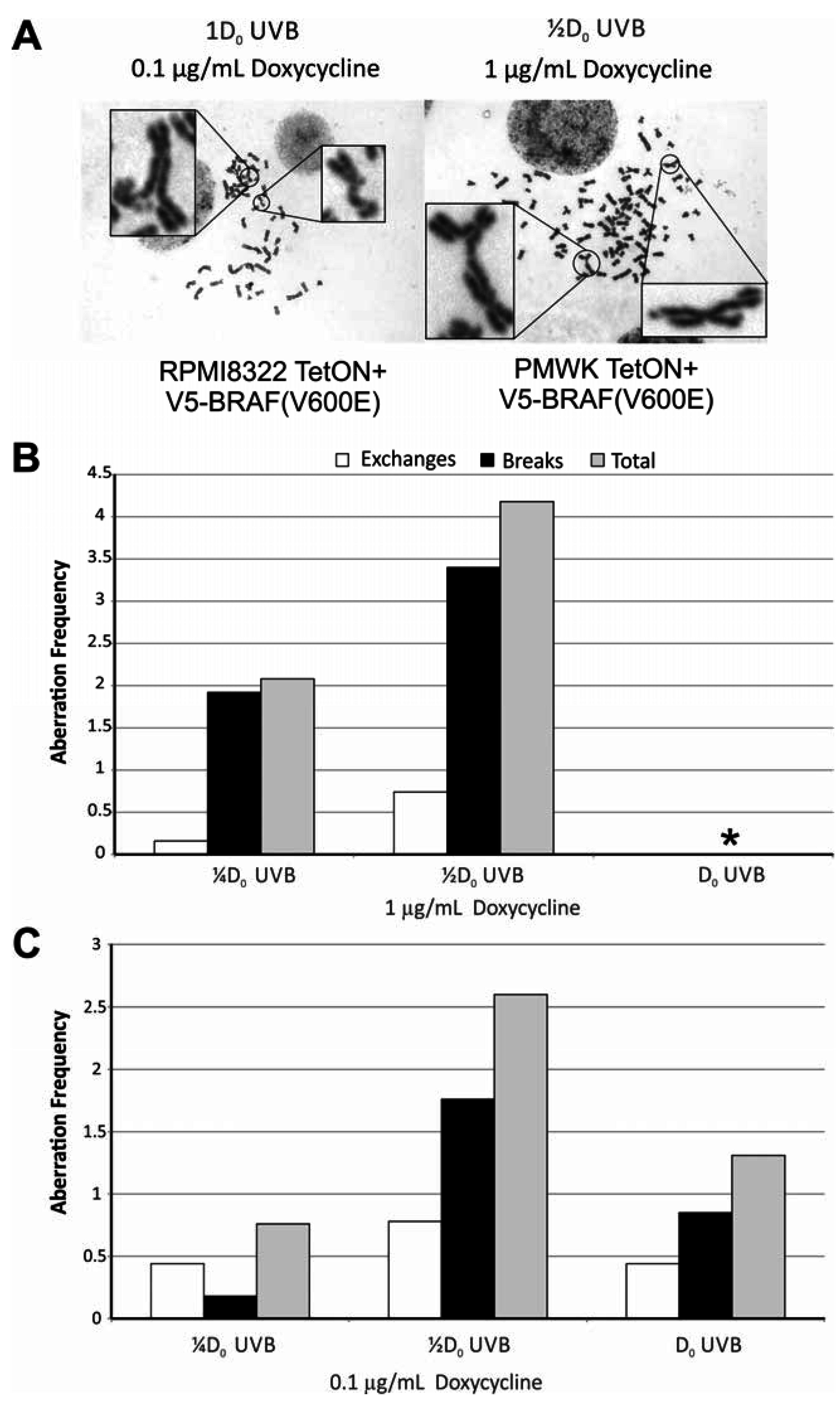
2.5. Oncogenic BRAF Sensitizes Cells to UVB Clastogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment/Sham | (Freq-Dox Freq)/Sham | ||||
|---|---|---|---|---|---|
| ¼D0 | ½D0 | * Dox | ¼D0 + * Dox | ½D0 + * Dox | |
| § PMWK | 4.5 | 16 | 7.3 | 10 | 28 |
| † RPMI8322 | 5.0 | 4.4 | 3.0 | 4.6 | 23 |
2.6. Mechanisms of BRAF(V600E) Induced Sensitivity
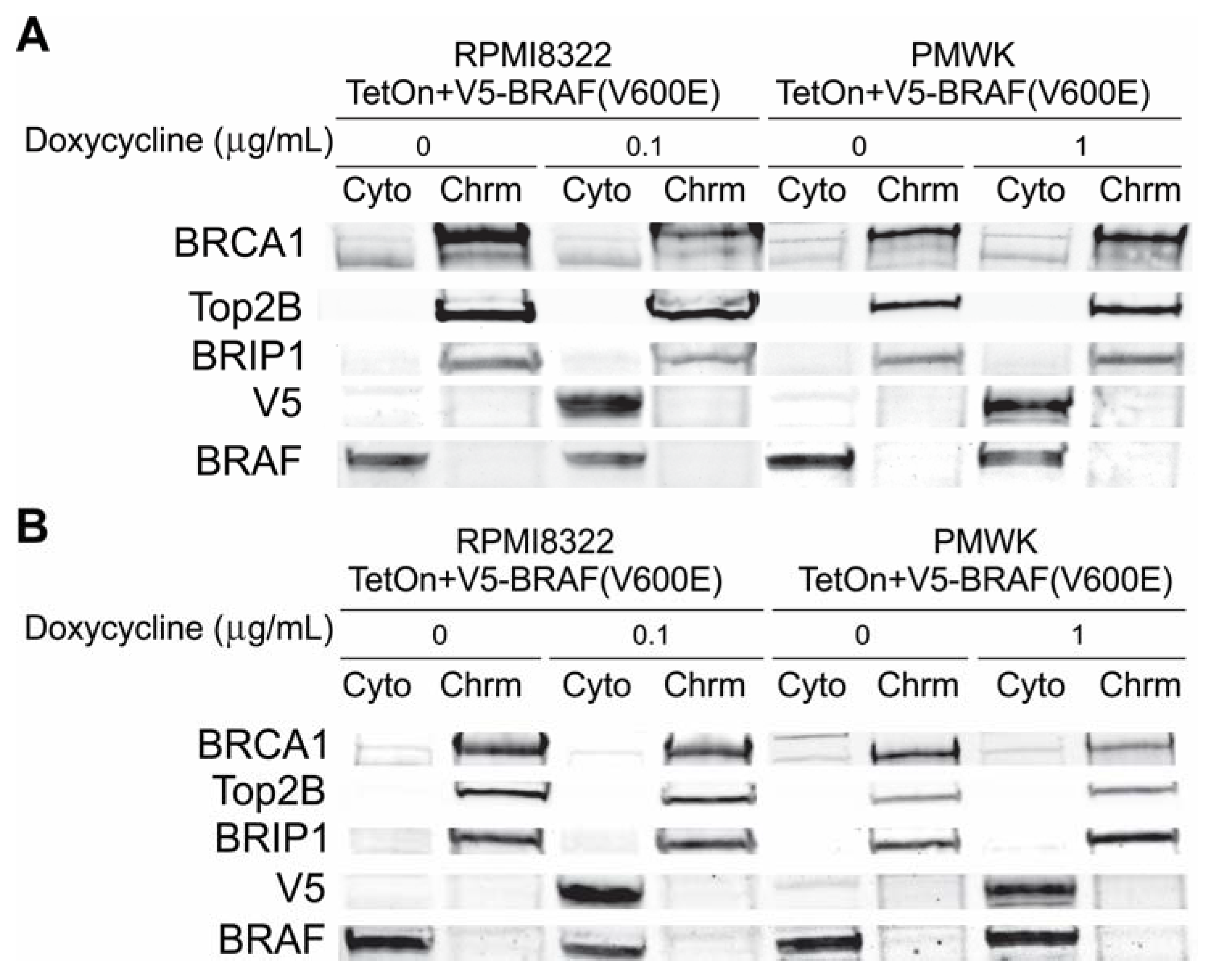
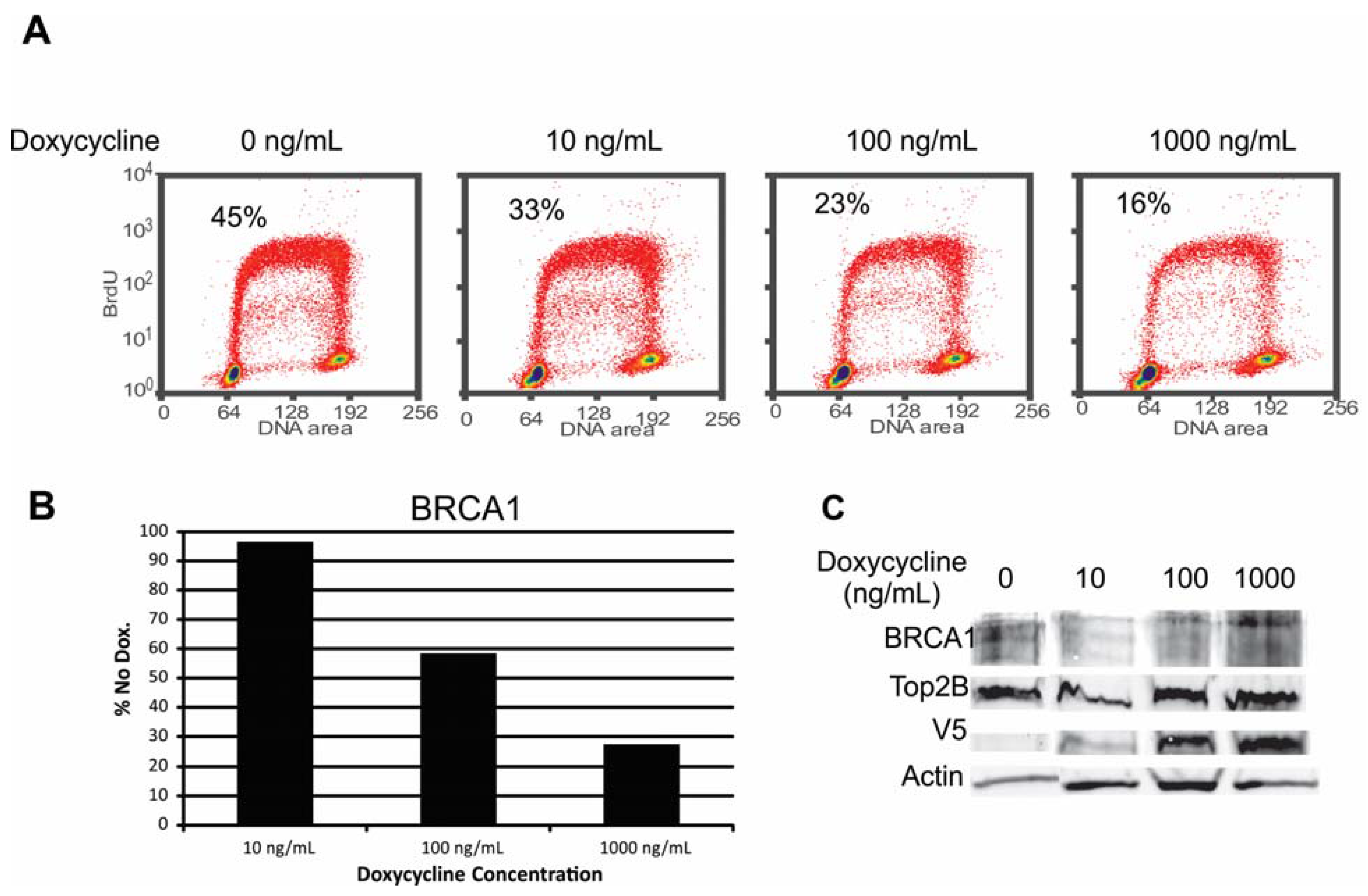
2.6.1. Alteration of BRCA1 Function


2.6.2. Alteration of Chk1 Signaling

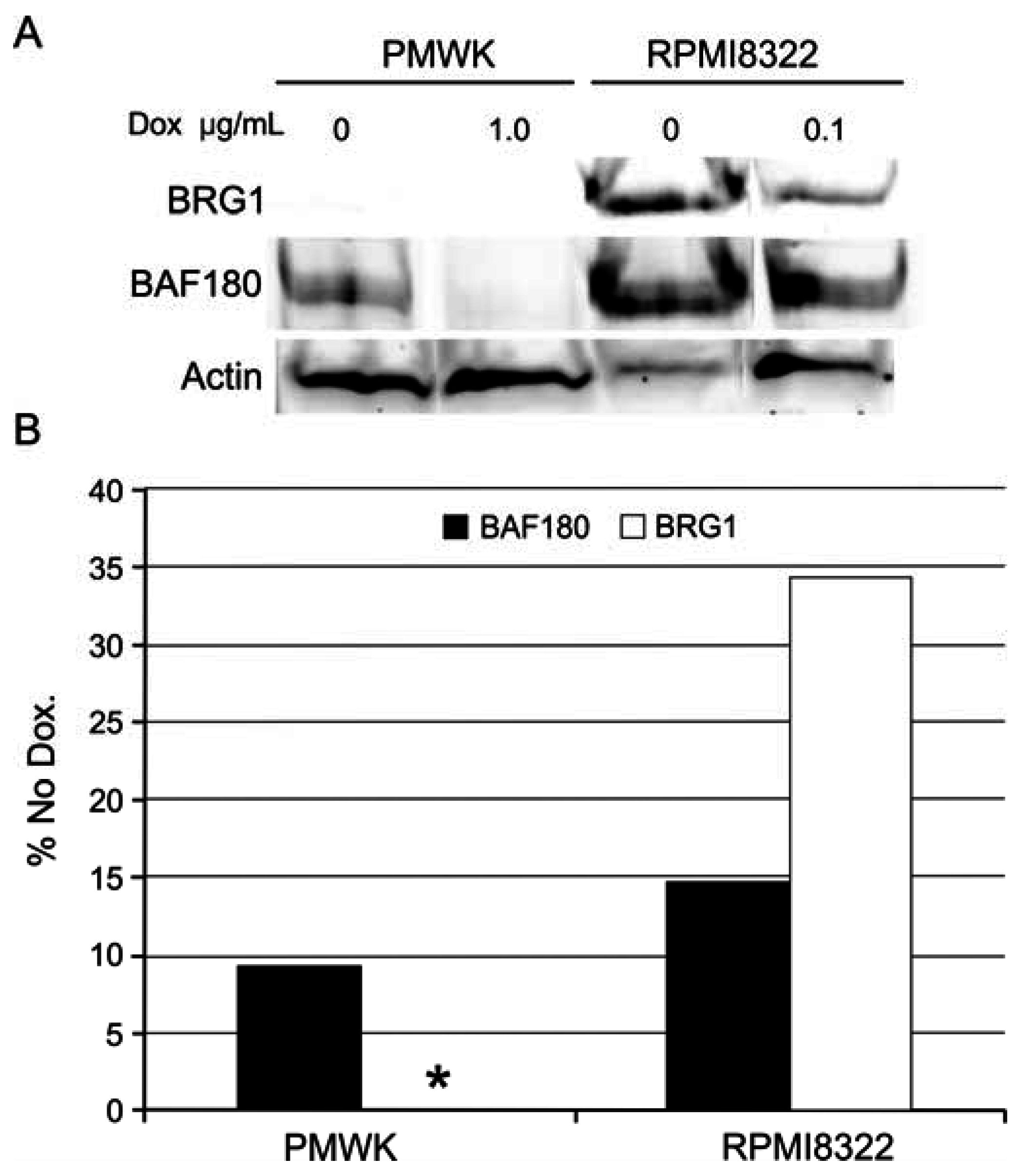
2.6.3. Alteration of SWI/SNF Chromatin Remodeling Complex

3. Experimental Section
3.1. Molecular Biology
3.2. Cell Culture and Viral Transduction
3.3. Cytotoxicity Assays
3.4. Western Blotting
| Antibody | Dilution | Source | Catalogue Number |
|---|---|---|---|
| BRCA1 | 1:1000 | Milli-Pore | 07-434 |
| Top2β | 1:1000 | BD Transduction Labs | 611493 |
| BRIP1/BACH1 | 1:1000 | Cell Signaling | 4578 |
| BRG1 | 1:1000 | Bethyl | A300-813A |
| BRAF | 1:1000 | Cell Signaling | 9434 |
| V5 | 1:1000 | Sigma-Aldrich | 012M4796 |
| Chk1 | 1:1000 | Santa Cruz | SC-8408 |
| pChk1 (S345) | 1:1000 | Cell Signaling | 2348 |
| pChk1 (S280) | 1:1000 | Epitomics | 2643-1 |
| Mek1/2 | 1:1000 | Cell Signaling | 4694 |
| pMek1/2 (S217/S221) | 1:1000 | Cell Signaling | 9154 |
| Pan-Actin | 1:5000 | Novus Biologics | NB600-535 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of braf mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Safaee Ardekani, G.; Jafarnejad, S.M.; Tan, L.; Saeedi, A.; Li, G. The prognostic value of braf mutation in colorectal cancer and melanoma: A systematic review and meta-analysis. PLoS ONE 2012, 7, e47054. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Millikan, R.C.; Groben, P.A.; Hao, H.; Tolbert, D.; Berwick, M.; Busam, K.; Begg, C.B.; et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 991. [Google Scholar] [CrossRef] [PubMed]
- Solus, J.F.; Kraft, S. Ras, Raf, and Map kinase in melanoma. Adv. Anat. Pathol. 2013, 20, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bianchi-Smiraglia, A.; Nikiforov, M.A. Controversial aspects of oncogene-induced senescence. Cell Cycle 2012, 11, 4147–4151. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.L.; Haferkamp, S.; Scurr, L.L.; Gowrishankar, K.; Becker, T.M.; Desilva, C.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F.; Rizos, H. Absence of distinguishing senescence traits in human melanocytic nevi. J. Investig. Dermatol. 2012, 132, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar] [PubMed]
- Bielas, J.H.; Loeb, L.A. Mutator phenotype in cancer: Timing and perspectives. Environ. Mol. Mutagen. 2005, 45, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, N.; Knauf, J.A.; Mitsutake, S.; Mesa, C., Jr.; Zhang, L.; Fagin, J.A. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid pccl3 cells. Cancer Res. 2005, 65, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.J.; Guan, B.; Tsai, F.J.; Hsiao, E.Y.; Chen, C.M.; Seruca, R.; Wang, T.L.; Shih, I.-M. Mutant braf induces DNA strand breaks, activates DNA damage response pathway, and up-regulates glucose transporter-1 in nontransformed epithelial cells. Am. J. Pathol. 2012, 180, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Aird, K.M.; Bitler, B.G.; Nicodemus, J.P.; Beeharry, N.; Xia, B.; Yen, T.J.; Zhang, R. Oncogenic Ras regulates brip1 expression to induce dissociation of brca1 from chromatin, inhibit DNA repair, and promote senescence. Dev. Cell. 2011, 21, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, M.; Rodriguez, J.A.; Baer, R.; Henderson, B.R. Bard1 induces brca1 intranuclear foci formation by increasing ring-dependent brca1 nuclear import and inhibiting brca1 nuclear export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar] [CrossRef] [PubMed]
- Omolo, B.; Carson, C.; Chu, H.; Zhou, Y.; Simpson, D.A.; Hesse, J.E.; Paules, R.S.; Nyhan, K.C.; Ibrahim, J.G.; Kaufmann, W.K. A prognostic signature of g(2) checkpoint function in melanoma cell lines. Cell Cycle 2013, 12, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.; Omolo, B.; Chu, H.; Zhou, Y.; Sambade, M.J.; Peters, E.C.; Tompkins, P.; Simpson, D.A.; Thomas, N.E.; Fan, C.; et al. A prognostic signature of defective p53-dependent g1 checkpoint function in melanoma cell lines. Pigment. Cell. Melanoma Res. 2012, 25, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K.; Nevis, K.R.; Qu, P.; Ibrahim, J.G.; Zhou, T.; Zhou, Y.; Simpson, D.A.; Helms-Deaton, J.; Cordeiro-Stone, M.; Moore, D.T.; et al. Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J. Investig. Dermatol. 2008, 128, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Sambade, M.J.; Peters, E.C.; Thomas, N.E.; Kaufmann, W.K.; Kimple, R.J.; Shields, J.M. Melanoma cells show a heterogeneous range of sensitivity to ionizing radiation and are radiosensitized by inhibition of B-RAF with PLX-4032. Radiother. Oncol. 2011, 98, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K.; Filatov, L.; Oglesbee, S.E.; Simpson, D.A.; Lotano, M.A.; McKeen, H.D.; Sawyer, L.R.; Moore, D.T.; Millikan, R.C.; Cordeiro-Stone, M.; et al. Radiation clastogenesis and cell cycle checkpoint function as functional markers of breast cancer risk. Carcinogenesis 2006, 27, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Gilchrest, B.A.; Eller, M.S.; Geller, A.C.; Yaar, M. The pathogenesis of melanoma induced by ultraviolet radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [PubMed]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates braf-driven melanomagenesis by targeting tp53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Marcus, P.I. Action of X-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Gaddameedhi, S.; Kemp, M.G.; Reardon, J.T.; Shields, J.M.; Smith-Roe, S.L.; Kaufmann, W.K.; Sancar, A. Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 2010, 70, 4922–4930. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Nguyen, J.; Hill, S.J.; Scully, R.; Adelmant, G.O.; Marto, J.A.; Feunteun, J.; Livingston, D.M. Brca1 is required for postreplication repair after UV-induced DNA damage. Mol. Cell. 2011, 44, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.S.; Nowsheen, S.; Rahman, M.A.; Cook, R.S.; Xia, F. Targeting brca1 localization to augment breast tumor sensitivity to poly(adp-ribose) polymerase inhibition. Cancer Res. 2012, 72, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002, 13, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chou, J.; Mullen, T.E.; Elkon, R.; Zhou, Y.; Simpson, D.A.; Bushel, P.R.; Paules, R.S.; Lobenhofer, E.K.; Hurban, P.; et al. Identification of primary transcriptional regulation of cell cycle-regulated genes upon DNA damage. Cell Cycle 2007, 6, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Parsons, R. Pten loss inhibits Chk1 to cause double stranded-DNA breaks in cells. Cell Cycle 2005, 4, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Goto, H.; Kasahara, K.; Matsuyama, M.; Wang, Z.; Yatabe, Y.; Kiyono, T.; Inagaki, M. P90 Rsk arranges Chk1 in the nucleus for monitoring of genomic integrity during cell proliferation. Mol. Biol. Cell. 2012, 23, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of pten sequesters Chk1 and initiates genetic instability. Cancer Cell. 2005, 7, 193–204. [Google Scholar] [CrossRef] [PubMed]
- King, F.W.; Skeen, J.; Hay, N.; Shtivelman, E. Inhibition of Chk1 by activated PKB/Akt. Cell Cycle 2004, 3, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Tardell, C.; Higgins, B.; Packman, K.; Boylan, J.F.; Niu, H. BRAFV600E negatively regulates the akt pathway in melanoma cell lines. PLoS ONE 2012, 7, e42598. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell. Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ray-David, H.; Romeo, Y.; Lavoie, G.; Deleris, P.; Tcherkezian, J.; Galan, J.A.; Roux, P.P. Rsk promotes g2 DNA damage checkpoint silencing and participates in melanoma chemoresistance. Oncogene 2013, 32, 4480–4489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, H.; Gong, M.; Gong, F. The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front. Oncol. 2013, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.; Heinonen, M.; Kantonen, J.; Gylling, A.; Lahtinen, L.; Korhonen, M.; Kytola, S.; Mecklin, J.P.; Orpana, A.; Peltomaki, P.; et al. BRAF mutation in sporadic colorectal cancer and lynch syndrome. Virchows Arch. 2013, 463, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.A.; Livanos, E.; Heffernan, T.P.; Kaufmann, W.K. Telomerase expression is sufficient for chromosomal integrity in cells lacking p53 dependent G1 checkpoint function. J. Carcinog. Electron. Resour. 2005, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.B.; Peritz, A.E.; Uitto, J.; Gasparro, F.P. Ultraviolet-filtering properties of commonly used tissue cell culture plasticware. Photodermatol. Photoimmunol. Photomed. 2001, 17, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.; Stillman, B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: Assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 2000, 20, 8602–8612. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Moussa, G.; Tomi, N.S.; Paech, V.; Altmeyer, P.; Kreuter, A. Reference limits for erythema-effective UV doses. Photochem. Photobiol. 2006, 82, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Smith-Roe, S.L.; Nakamura, J.; Holley, D.; Chastain, P.D., 2nd; Rosson, G.B.; Simpson, D.A.; Ridpath, J.R.; Kaufman, D.G.; Kaufmann, W.K.; Bultman, S.J. SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget 2015, 6, 732–745. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simpson, D.A.; Lemonie, N.; Morgan, D.S.; Gaddameedhi, S.; Kaufmann, W.K. Oncogenic BRAF(V600E) Induces Clastogenesis and UVB Hypersensitivity. Cancers 2015, 7, 1072-1090. https://doi.org/10.3390/cancers7020825
Simpson DA, Lemonie N, Morgan DS, Gaddameedhi S, Kaufmann WK. Oncogenic BRAF(V600E) Induces Clastogenesis and UVB Hypersensitivity. Cancers. 2015; 7(2):1072-1090. https://doi.org/10.3390/cancers7020825
Chicago/Turabian StyleSimpson, Dennis A., Nathalay Lemonie, David S. Morgan, Shobhan Gaddameedhi, and William K. Kaufmann. 2015. "Oncogenic BRAF(V600E) Induces Clastogenesis and UVB Hypersensitivity" Cancers 7, no. 2: 1072-1090. https://doi.org/10.3390/cancers7020825
APA StyleSimpson, D. A., Lemonie, N., Morgan, D. S., Gaddameedhi, S., & Kaufmann, W. K. (2015). Oncogenic BRAF(V600E) Induces Clastogenesis and UVB Hypersensitivity. Cancers, 7(2), 1072-1090. https://doi.org/10.3390/cancers7020825