
High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer

Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.2. Experimental Diets
2.3. Experimental Procedure
2.4. Primary Tumor Volume Measurements
2.5. Lung Metastases
2.6. Hepatic Steatosis
2.7. Infiltrating Macrophages and Microvessel Densities
2.8. Crown-Like Structures
2.9. Plasma Cytokines and Tissue MCP-1
2.10. Statistical Analyses
3. Results
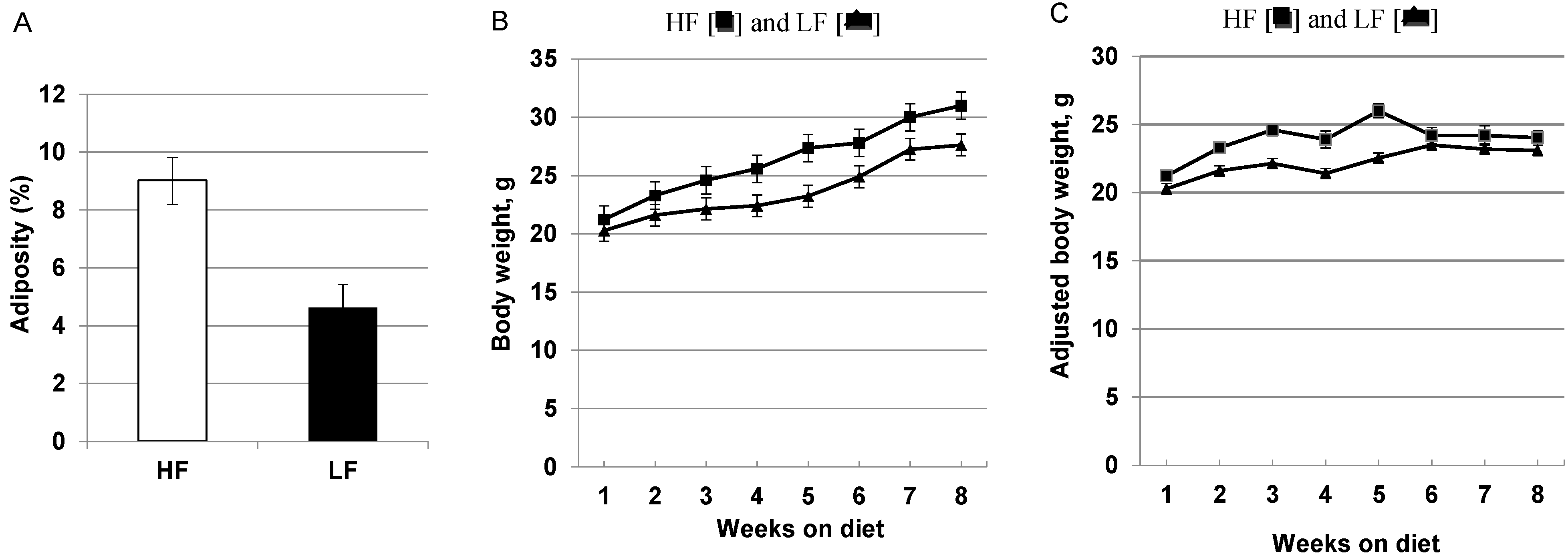
3.1. High-Fat, High Calorie Diet Increases Adiposity, Weight Gain and Hepatic Steatosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4 weeks | Absent | Trace | Mild | Moderate | p-value * | |||||
| n | % | n | % | n | % | n | % | |||
| LF | 1 | 16.67 | 2 | 33.33 | 3 | 50.00 | 0 | 0.00 | 0.435 | |
| HF | 1 | 16.67 | 4 | 66.67 | 1 | 16.67 | 0 | 0.00 | ||
| 8 weeks | Absent | Trace | Mild | Moderate | p-value * | |||||
| n | % | n | % | n | % | n | % | |||
| LF | 3 | 21.43 | 8 | 57.14 | 3 | 21.43 | 0 | 0.00 | <0.0001 | |
| HF | 0 | 0.00 | 0 | 0.00 | 11 | 78.57 | 3 | 21.43 | ||
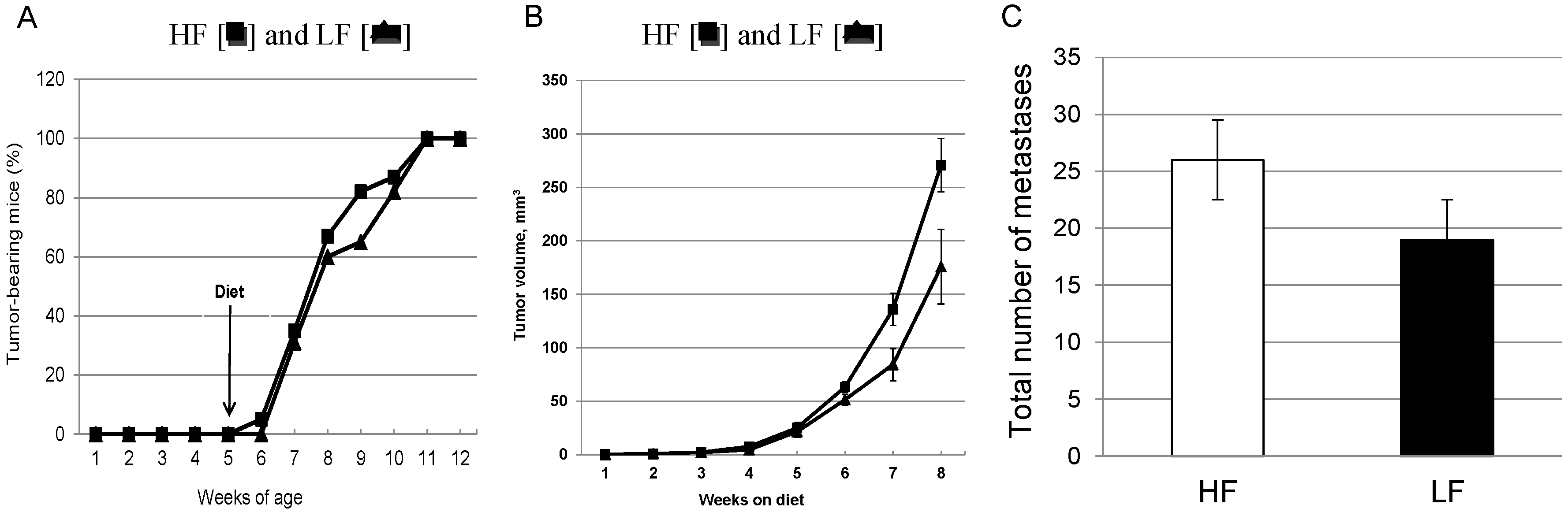
3.2. Chronic Consumption of a High-Fat Diet Significantly Increases Primary Mammary Tumor Growth without Effects on Latency in Female MMTV-PyMT Mice

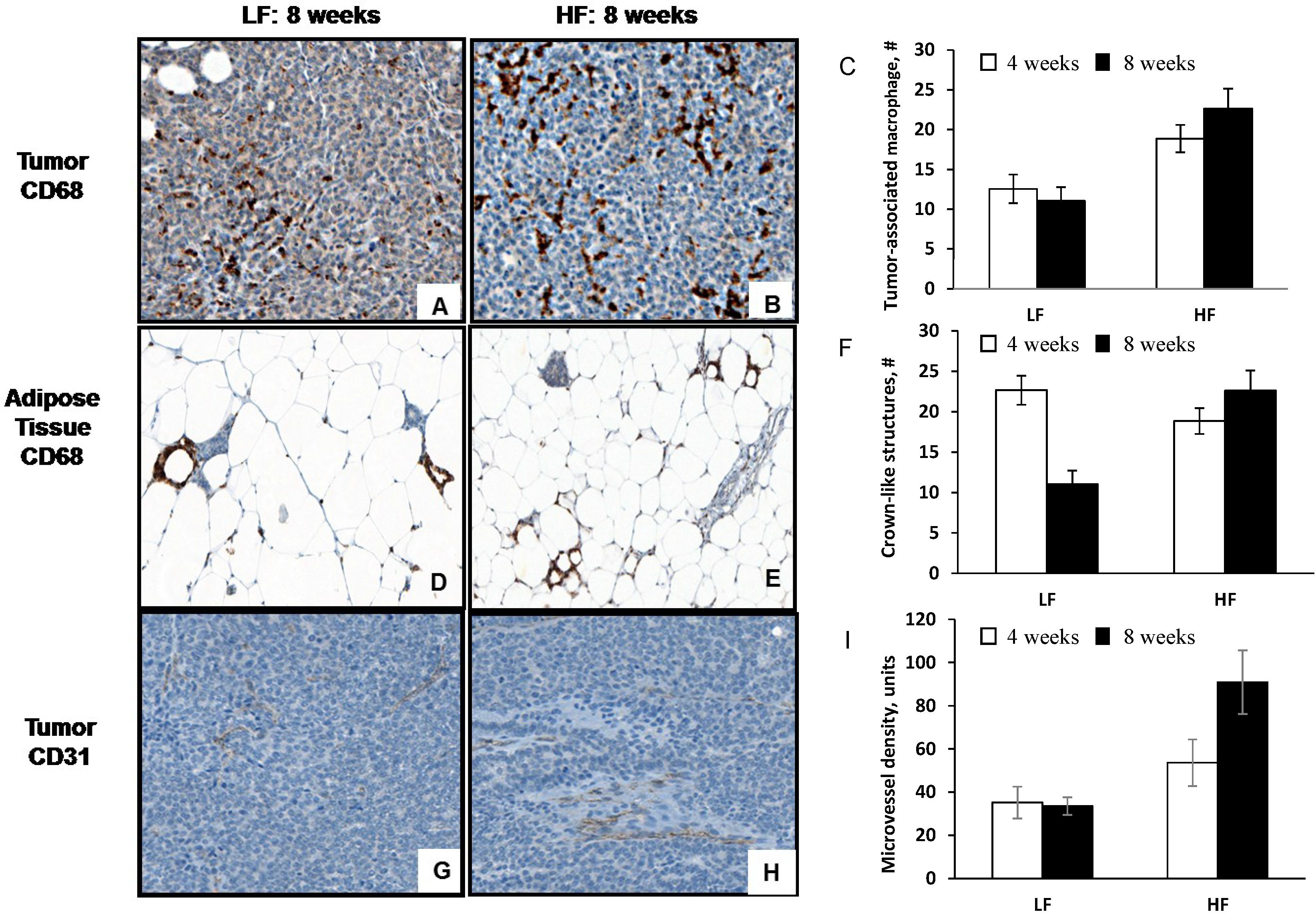
3.3. Diet-Induced Obesity Increases Tumor-Associated Macrophage Infiltration, Crown-Like Structures and Microvessel Density

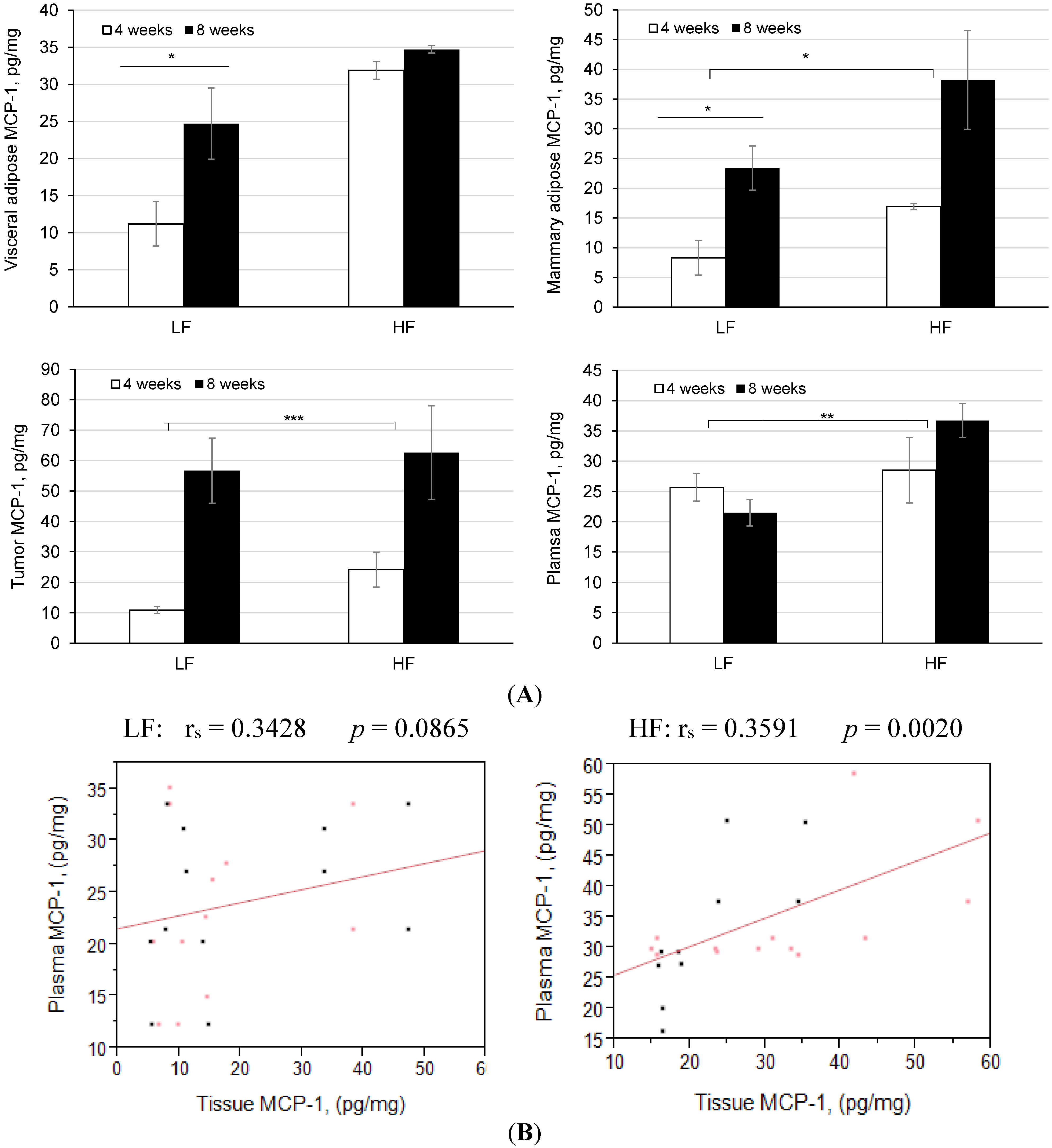
3.4. Diet-Induced Obesity and Inflammation Significantly Increases Plasma Cytokines and Mammary Tumor Mcp-1 Production
| Plasma Protein | Units | HF | LF | p-value |
|---|---|---|---|---|
| TNF-α | pg/mL | 11.8 ± 2.0 | 7.5 ± 1.0 | 0.246 |
| IL-6 | pg/mL | 3.0 ± 0.1 | 1.5 ± 0.2 | 0.059 |
| IL-10 | pg/mL | 20.1 ± 4.0 | 14.0 ± 0.2 | 0.410 |
| MCP-1 | pg/mL | 38.0 ± 0.3 | 22.0 ± 0.2 | 0.002 |
| Leptin | ng/mL | 8.3 ± 1.4 | 2.1 ± 0.5 | <0.050 |

4. Discussion
5. Concluding Remarks and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rinaldi, S.; Key, T.J.; Peeters, P.H.; Lahmann, P.H.; Lukanova, A.; Dossus, L.; Biessy, C.; Vineis, P.; Sacerdote, C.; Berrino, F.; et al. Anthropometric measures, endogenous sex steroids and breast cancer risk in postmenopausal women: a study within the EPIC cohort. Int. J. Cancer 2006, 118, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. Interaction between menopausal status and obesity in affecting breast cancer risk. Maturitas 2010, 66, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Rohan, T.E.; Heo, M.; Choi, L.; Datta, M.; Fredenheim, J.L.; Kamensky, V.; Ochs-Balcom, H.M.; Qi, L.; Thomson, C.A.; Vitolins, M.Z.; et al. Body fat and breast cancer risk in postmenopausal women: a longitudinal study. J. Cancer Epidemiol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. Influence of obesity on breast cancer receptor status and prognosis. Expert. Rev. Anticancer Ther. 2009, 9, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Stulnig, T.M. Obesity, inflammation, and insulin resistance—A mini-review. Gerontology 2009, 55, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Manabe, I. Chronic inflammation links cardiovascular, metabolic, and renal diseases. Circ. J. 2011, 75, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Okwan-Duodu, D.; Umpierrez, G.E.; Brawley, O.W.; Diaz, R. Obesity-driven inflammation, and cancer risk: role of myeloid derived suppressor cells and alternately activated macrophages. Am. J. Cancer Res. 2013, 3, 21–33. [Google Scholar] [PubMed]
- Vona-Davis, L.; Rose, D.P. The Obesity-inflammation-eicosanoid axis in breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desi, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Mumick, S.; Zhang, C.; Lamb, J.; Dai, H.; Weingarth, D.; Mudgett, J.; Chen, H.; MacNeil, D.J.; Reitman, M.L.; et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 2005, 13, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jenkins, J.R.; Trayhum, P. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: integrated response to TNF-alpha. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mizuarai, S.; Araki, H.; Maskiko, S.; Ishihara, A.; Kanatani, A.; Itadani, H.; Kotani, H. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. J. Biol. Chem. 2003, 278, 46654–46660. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer. Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 2009, 20, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Tahergorabi, Z.; Khazaei, M. The relationship between inflammatory markers, angiogenesis, and obesity. ARYA Atheroscler 2013, 9, 247–253. [Google Scholar] [PubMed]
- Yoshimura, T.; Howard, O.M.; Ito, T.; Kuwabara, M.; Matsukawa, A.; Chen, K.; Liu, Y.; Liu, M.; Oppenheim, J.J.; Wang, J.M. Monocyte chemoattractant protein-1/CCL2 produced by stromal cells promotes lung metastasis of 4T1 murine breast cancer cells. PLoS ONE 2013, 8, e58791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brakenhielm, E.; Cao, R.; Gao, B.; Angelin, B.; Cannon, B.; Parini, P.; Cao, Y. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ. Res. 2004, 94, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ono, M.; Shono, T.; Izumi, H.; Ishibashi, T.; Suzuki, H.; Kuwano, M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor α-dependent angiogenesis. Mol. Cell. Biol. 1997, 17, 4015–4023. [Google Scholar] [PubMed]
- Williams, J.C.; Gusterson, B.; Humphreys, J.; Monaghan, P.; Coombes, R.C.; Rudland, P.; Neville, A.M. N-methyl-N-nitrosourea-induced rat mammary tumors. Hormone responsiveness but lack of spontaneous metastasis. J. Natl. Cancer Inst. 1981, 66, 147–155. [Google Scholar] [PubMed]
- Stoica, G.; Koestner, A.; Capen, C.C. Neoplasms induced with high single doses of N-ethyl-N-nitrosourea in 30-day-old Sprague-Dawley rats, with special emphasis on mammary neoplasia. Anticancer Res. 1984, 4, 5–12. [Google Scholar] [PubMed]
- Rose, D.P.; Connolly, J.M. Dietary fat and breast cancer metastasis by human tumor xenografts. Breast Cancer Res. Treat. 1997, 46, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Gordon, R.R.; Hunter, K.W.; La Merrill, M.; Sorensen, P.; Threadgill, D.W.; Pomp, D. Genotype X diet interactions in mice predisposed to mammary cancer. I. Body weight and fat. Mamm. Genome 2008, 19, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.R.; Hunter, K.W.; Sorensen, P.; Pomp, D. Genotype X diet interactions in mice predisposed to mammary cancer: II. Tumors and metastasis. Mamm. Genome 2008, 19, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Molnar, J.; Yu, S.; Mzhavia, N.; Pau, C.; Chereshney, I.; Dansky, H.M. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ. Res. 2005, 96, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Qing, K.; Chen, Y. Diet-induced changes in stearoyl-CoA desaturase 1 expression in obesity-prone and -resistant mice. Obes. Res. 2004, 12, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Lamping, K.G.; Nuno, D.W.; Coppey, L.J.; Holmes, A.J.; Hu, S.; Oltman, C.L.; Norris, A.W.; Yorek, M.A. Modification of high saturated fat diet with n-3 polyunsaturated fat improves glucose intolerance and vascular dysfunction. Diabetes Obes. Metab. 2013, 15, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, H.C.; Chalasani, N. Nonalcoholic fatty liver disease: An emerging threat to obese and diabetic individuals. Ann. N. Y. Acad. Sci. 2013, 1281, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K. Metabolic syndrome and postmenopausal breast cancer: Systematic review and meta-analysis. Menopause 2013, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Wildman, R.P.; Kaplan, R.; Manson, J.E.; Rajkovic, A.; Connelly, S.A.; Mackey, R.H.; Tinker, L.F.; Curb, J.D.; Eaton, C.B.; Wassertheil-Smoller, S. Body size phenotypes and inflammation in the Women’s Health Initiative Observational Study. Obesity 2011, 19, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L.; et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- McCune, K.; Mehta, R.; Thorat, M.A.; Badve, S.; Nakshatri, H. Loss of ERalpha and FOXA1 expression in a progression model of luminal type breast cancer: insights from PyMT transgenic mouse model. Oncol. Rep. 2010, 24, 1233–1239. [Google Scholar] [PubMed]
- Wagner, M.; Bjerkvig, R.; Wiig, H.; Melero-Martin, J.M.; Lin, R.Z.; Klagsbrun, M.; Dudley, A.C. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis 2012, 15, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Thomas, T.C.; Storlien, L.H.; Huang, X.F. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.; Yang, G.; Lee, I.; Bielawski, J.; Hannun, Y.A.; Samad, F. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J. Biol. Chem. 2008, 283, 13538–13548. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr. Opin. Hematol. 2010, 17, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Santander, A.M.; Lopez-Ocejo, O.; Casas, O.; Agostini, T.; Sanchez, L.; Lamas-Basulto, E.; Carrio, R.; Cleary, M.P.; Gonzalez-Perez, R.R.; Torroella-Kouri, M. Paracrine interactions between adipocytes and tumor cells recruit and modify macrophages to the mammary tumor microenvironment: The role of obesity and inflammation in breast adipose tissue. Cancers 2015, 7, 143–178. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cowen, S.; McLaughlin, S.L.; Hobbs, G.; Coad, J.; Martin, K.H.; Olfert, I.M.; Vona-Davis, L. High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer. Cancers 2015, 7, 1125-1142. https://doi.org/10.3390/cancers7030828
Cowen S, McLaughlin SL, Hobbs G, Coad J, Martin KH, Olfert IM, Vona-Davis L. High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer. Cancers. 2015; 7(3):1125-1142. https://doi.org/10.3390/cancers7030828
Chicago/Turabian StyleCowen, Sarah, Sarah L. McLaughlin, Gerald Hobbs, James Coad, Karen H. Martin, I. Mark Olfert, and Linda Vona-Davis. 2015. "High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer" Cancers 7, no. 3: 1125-1142. https://doi.org/10.3390/cancers7030828
APA StyleCowen, S., McLaughlin, S. L., Hobbs, G., Coad, J., Martin, K. H., Olfert, I. M., & Vona-Davis, L. (2015). High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer. Cancers, 7(3), 1125-1142. https://doi.org/10.3390/cancers7030828