
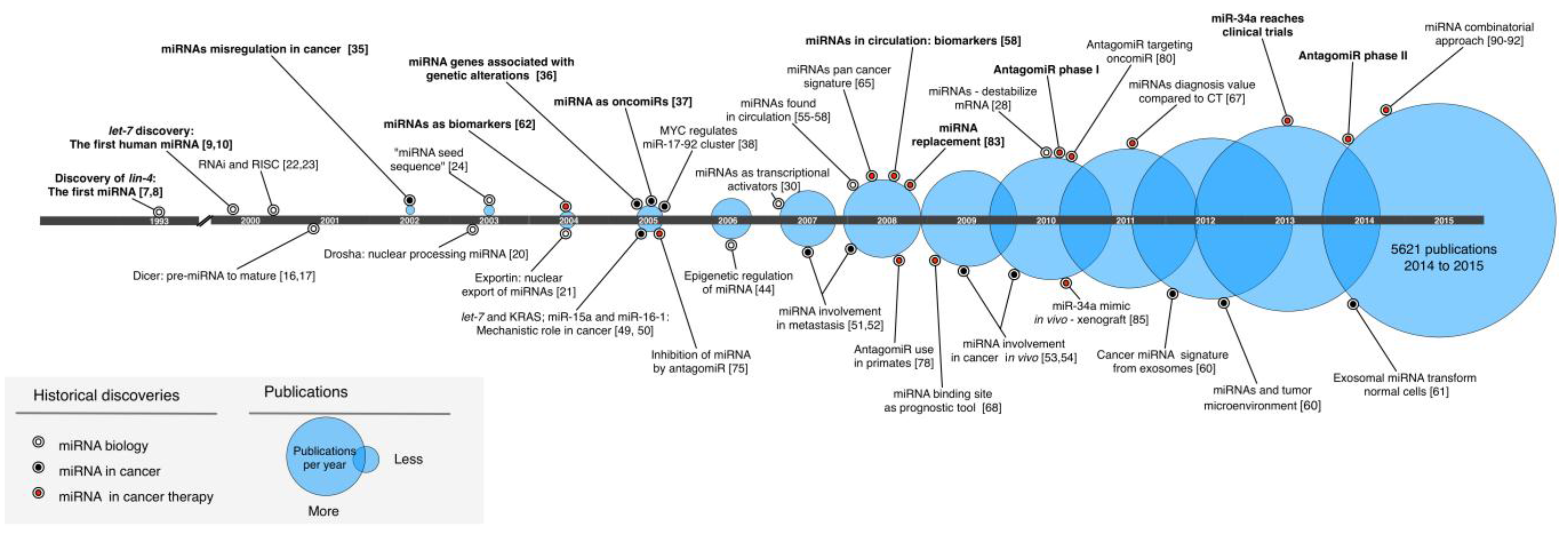
MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy

{kind=link}
Abstract
:1. Introduction
2. Early History: MiRNA Discovery

3. MiRNA Basic Biology
3.1. MiRNA Biogenesis
3.2. MiRNA Biological Function
4. MiRNA Misregulation in Cancer
5. MiRNA Transition into Therapy
5.1. Diagnosis, Detection and Prognosis
5.2. MiRNAs as Therapeutic Agents
6. Closing Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, R.; Feinbaum, R.; Ambros, V. A short history of a short RNA. Cell 2004, 116, S89–S92. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Horvitz, H.R.; Sulston, J.E. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics 1980, 96, 435–454. [Google Scholar] [PubMed]
- Chalfie, M.; Horvitz, H.R.; Sulston, J.E. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 1981, 24, 59–69. [Google Scholar] [CrossRef]
- Ambros, V.; Horvitz, H.R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, E.L.; Sternberg, P.W.; Horvitz, H.R. A genetic pathway for the specification of the vulval cell lineages of Caenorhabditis elegans. Nature 1987, 326, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [PubMed]
- Slack, F.J.; Basson, M.; Liu, Z.; Ambros, V.; Horvitz, H.R.; Ruvkun, G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell 2000, 5, 659–669. [Google Scholar] [CrossRef]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–964. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs that Control C. elegans Developmental Timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; Kim, V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [PubMed]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5ʹ UTR as in the 3ʹ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J.; et al. MiR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010, 140, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. P53-Mediated Activation of miRNA34 Candidate Tumor-Suppressor Genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Körner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. MiR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alarcón, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massagué, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Costinean, S.; Sandhu, S.K.; Pedersen, I.M.; Tili, E.; Trotta, R.; Perrotti, D.; Ciarlariello, D.; Neviani, P.; Harb, J.; Kauffman, L.R.; et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood 2009, 114, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.P.; Nolde, M.; Slack, F.J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Chim, S.S.C.; Shing, T.K.F.; Hung, E.C.W.; Leung, T.Y.; Lau, T.K.; Chiu, R.W.K.; Lo, Y.M.D. Detection and characterization of placental microRNAs in maternal plasma. Clin. Chem. 2008, 54, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, C.H.; Gal, S.; Dunlop, H.M.; Pushkaran, B.; Liggins, A.P.; Pulford, K.; Banham, A.H.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008, 141, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.P.; Rajapakshe, K.; Hartig, S.M.; Reva, B.; McLellan, M.D.; Kandoth, C.; Ding, L.; Zack, T.I.; Gunaratne, P.H.; Wheeler, D.A.; et al. Identification of a pan-cancer oncogenic microRNA superfamily anchored by a central core seed motif. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Gahan, P.B. Circulating nucleic acids in plasma and serum: Applications in diagnostic techniques for noninvasive prenatal diagnosis. Int. J. Womens. Health 2013, 5, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.U.; Straka, E.; Su, L.; Burki, E.A.; et al. A SNP in a let-7 microRNA complementary site in the KRAS 3ʹ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008, 68, 8535–8840. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Weidhaas, J.B.; Lenz, H.J.; Laurent-Puig, P.; Jacobs, B.; De Schutter, J.; De Roock, W.; Salzman, D.W.; Zhang, W.; Yang, D.; et al. A let-7 microRNA-binding site polymorphism in KRAS predicts improved outcome in patients with metastatic colorectal cancer treated with salvage cetuximab/panitumumab monotherapy. Clin. Cancer Res. 2014, 20, 4499–4510. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Lee, A.M.; Shi, Q.; Alberts, S.R.; Sargent, D.J.; Sinicrope, F.A.; Diasio, R.B. Association study of the let-7 miRNA-complementary site variant in the 3ʹ untranslated region of the KRAS gene in stage III colon cancer (NCCTG N0147 Clinical Trial). Clin. Cancer Res. 2014, 20, 3319–3327. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Lee, J.W.; Slebos, R.J.; Howard, J.D.; Perez, J.; Kang, H.; Fertig, E.J.; Considine, M.; Gilbert, J.; Murphy, B.A.; et al. A 3ʹ-UTR KRAS-variant is associated with cisplatin resistance in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2014, 25, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Bueno Santiago, M.; de Lima Marson, F.A.; Secolin, R.; Ribeiro, J.D.; Passos Lima, C.S.; Bertuzzo, C.S. SLC23A2-05 (rs4987219) and KRAS‑LCS6 (rs61764370) polymorphisms in patients with squamous cell carcinoma of the head and neck. Oncol. Lett. 2014, 7, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.J.; Ratner, E.; Uduman, M.; Winter, K.; Boeke, M.; Greven, K.M.; King, S.; Burke, T.W.; Underhill, K.; Kim, H.; et al. The KRAS-variant and miRNA expression in RTOG endometrial cancer clinical trials 9708 and 9905. PLoS ONE 2014, 9, e94167. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R.; Patel, D.A.; Weitzel, J.; McVeigh, T.; Dorairaj, J.J.; Heneghan, H.M.; Miller, N.; Weidhaas, J.B.; Kerin, M.J.; McKenna, M.; et al. The KRAS-variant is associated with risk of developing double primary breast and ovarian cancer. PLoS ONE 2012, 7, e37891. [Google Scholar] [CrossRef] [PubMed]
- Cerne, J.Z.; Stegel, V.; Gersak, K.; Novakovic, S. KRAS rs61764370 is associated with HER2-overexpressed and poorly-differentiated breast cancer in hormone replacement therapy users: A case control study. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Hutvágner, G.; Simard, M.J.; Mello, C.C.; Zamore, P.D. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004, 2, e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.F.R.; Rebhan, M.A.E.; Crivelli, S.E.M.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014, 42, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2014, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. SiRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Slack, F.J. MiRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef] [PubMed]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Borad, M.; Sachdev, J.; Hong, D.S.; Smith, S.; Bader, A.; Stoudemire, J.; Kim, S.; Brenner, A. Abstract CT327: Multicenter phase I study of MRX34, a first-in-class microRNA miR-34 mimic liposomal injection. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Kelnar, K.; Stahlhut, C.; Orellana, E.; Zhao, J.; Shimer, E.; Dysart, S.; Chen, X.; Bader, A.G.; Slack, F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene 2015, 34, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Dahlman, J.E.; Tammela, T.; Khan, O.F.; Sood, S.; Dave, A.; Cai, W.; Chirino, L.M.; Yang, G.R.; Bronson, R.; et al. Small RNA combination therapy for lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3553–E3561. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, C.; Slack, F.J. Combinatorial action of MicroRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation. Cell Cycle 2015, 14, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. Next generation gene delivery approaches: Recent progress and hurdles. Mol. Pharm. 2015, 12, 299–300. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orellana, E.A.; Kasinski, A.L. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers 2015, 7, 1388-1405. https://doi.org/10.3390/cancers7030842
Orellana EA, Kasinski AL. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers. 2015; 7(3):1388-1405. https://doi.org/10.3390/cancers7030842
Chicago/Turabian StyleOrellana, Esteban A., and Andrea L. Kasinski. 2015. "MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy" Cancers 7, no. 3: 1388-1405. https://doi.org/10.3390/cancers7030842
APA StyleOrellana, E. A., & Kasinski, A. L. (2015). MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers, 7(3), 1388-1405. https://doi.org/10.3390/cancers7030842