
Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer?

Abstract
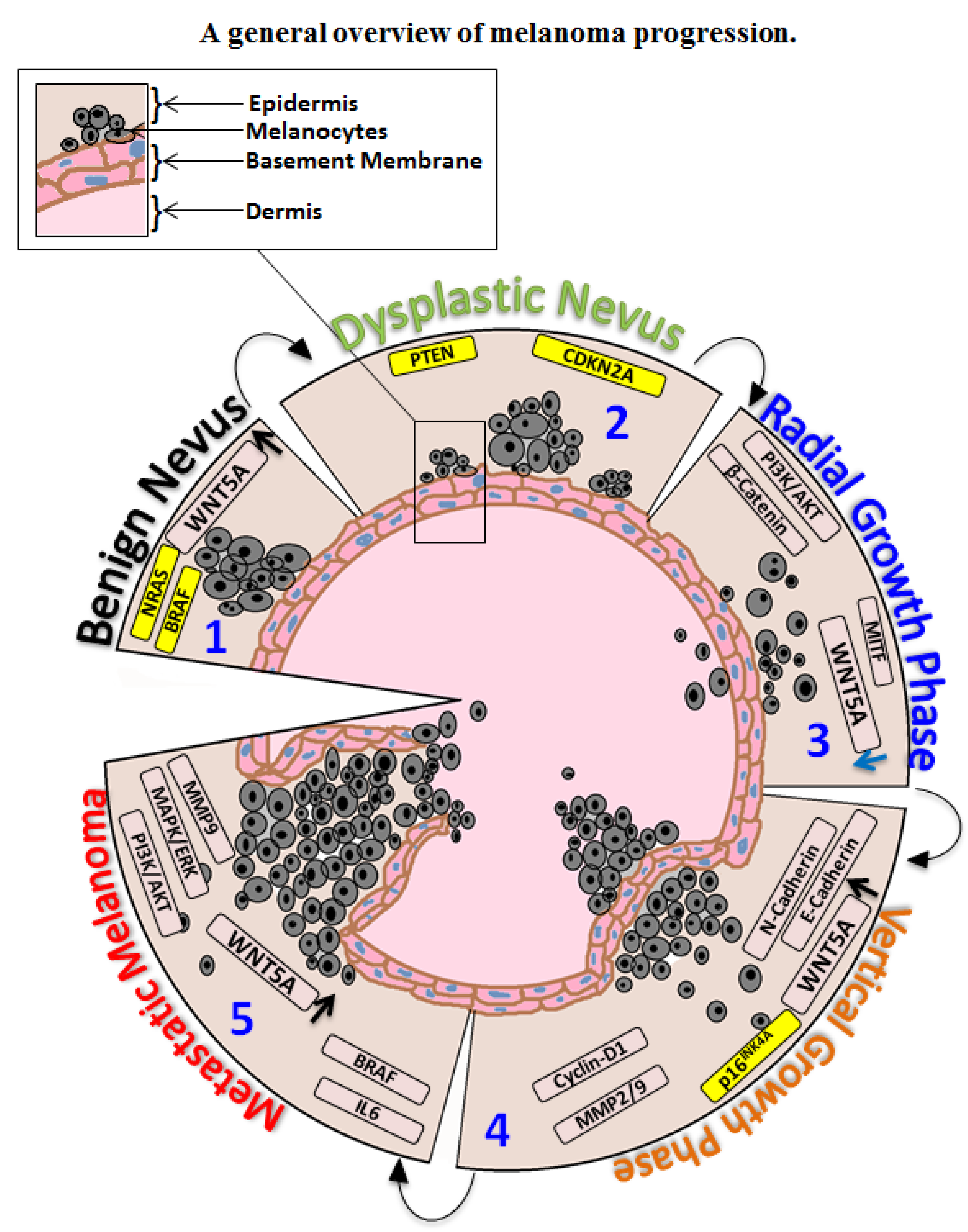
:1. Introduction
2. Oncogenic Signaling in Melanoma
2.1. RAS/RAF/MEK/ERK Signaling

2.2. The PI3K/AKT Pathway
2.3. WNT Signaling
2.3.1. WNT/β-Catenin Pathway
MITF
2.3.2. WNT5A Signaling
WNT5A Localization: A Topic of Debate
3. BRAF-Targeted Therapy in Melanoma
Signaling Pathways Responsible for Acquired Resistance to BRAFi
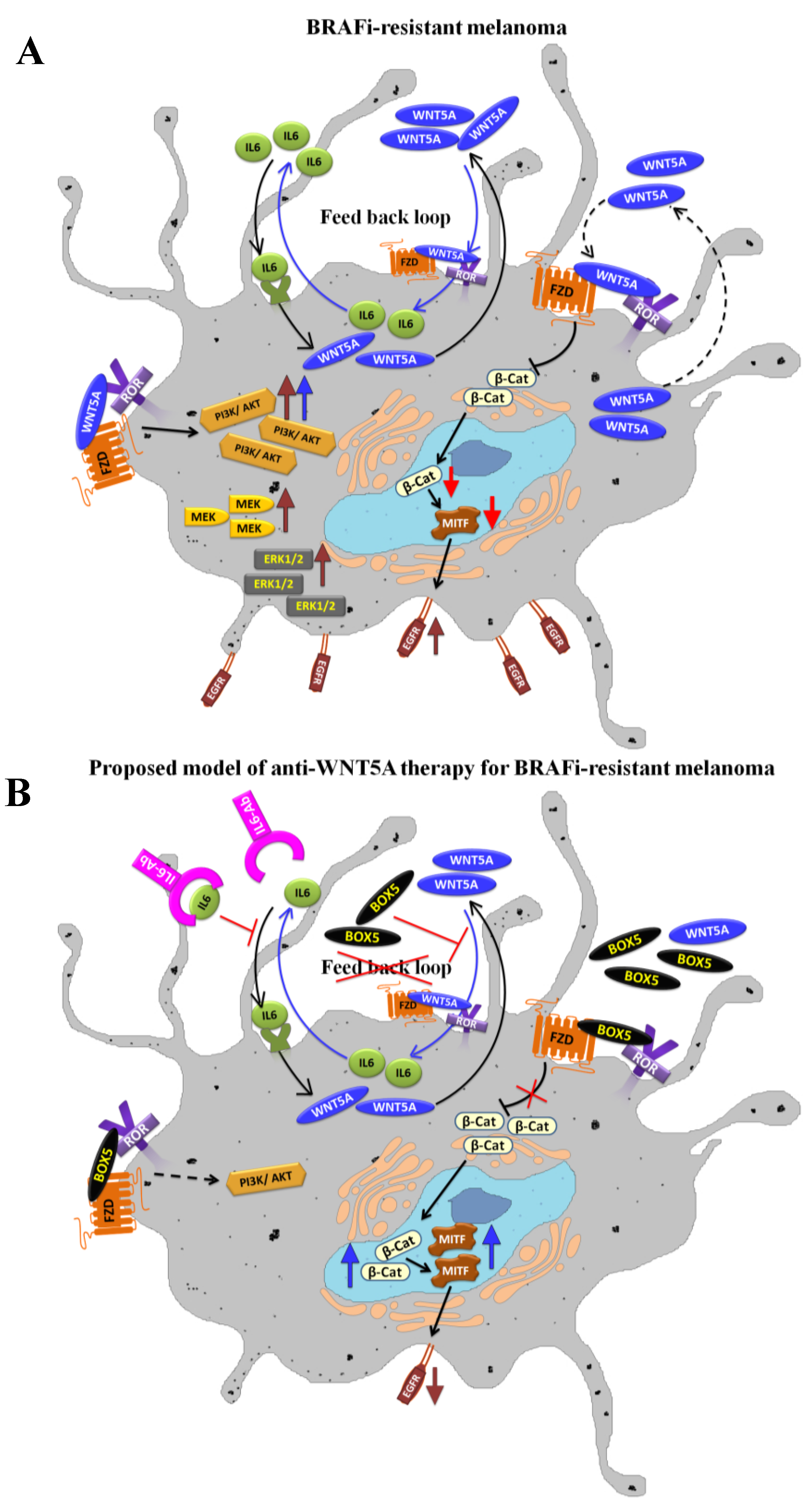
4. WNT5A Signaling in BRAFi-Resistance and Therapeutic Interventions


{kind=link}
{kind=link}
| Factors | Signaling Involved | References |
|---|---|---|
| BRAFV600E copy number amplification /or alternative splicing | MAPK signaling | [92,114,115,116,117] |
| MEK1/2 activating mutations | MAPK signaling | [118,119,120,121] |
| NRAS activating mutations | MAPK signaling | [91,92,116,121,122] |
| PTEN loss | PI3K/AKT signaling | [40,121,123] |
| Mutations in PI3K/AKT pathway e.g., AKT1/3, PI3KCA etc. | PI3K/AKT signaling | [92,117] |
| MITF downregulation | EGFR signaling | [98,124] |
| WNT5A up-regulation | Non-canonical WNT signaling | [99,100,101] |
5. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Gilchrest, B.A.; Eller, M.S. DNA photodamage stimulates melanogenesis and other photoprotective responses. J. Investig. Dermatol. Symp. Proc. 1999, 4, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Naysmith, L.; Waterston, K.; Ha, T.; Flanagan, N.; Bisset, Y.; Ray, A.; Wakamatsu, K.; Ito, S.; Rees, J.L. Quantitative measures of the effect of the melanocortin 1 receptor on human pigmentary status. J. Investig. Dermatol. 2004, 122, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P.; Healy, E.; Jackson, I.; Rees, J.L.; Thody, A.J. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet. 1995, 11, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.; ter Huurne, J.; Berkhout, M.; Gruis, N.; Bastiaens, M.; Bergman, W.; Willemze, R.; Bavinck, J.N. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J. Investig. Dermatol. 2001, 117, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Cutaneous melanoma. Lancet 2005, 365, 687–701. [Google Scholar] [CrossRef]
- Clark, W.H., Jr.; Elder, D.E.; Guerry, D.t.; Epstein, M.N.; Greene, M.H.; van Horn, M. A study of tumor progression: The precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984, 15, 1147–1165. [Google Scholar] [CrossRef]
- Breslow, A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann. Surg. 1970, 172, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin. Cancer Res. 2003, 9, 6483–6488. [Google Scholar] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human NAEVI. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Bevona, C.; Goggins, W.; Quinn, T.; Fullerton, J.; Tsao, H. Cutaneous melanomas associated with nevi. Arch. Dermatol. 2003, 139, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: A population-based estimate. Arch. Dermatol. 2003, 139, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Rizos, H. Human nevi lack distinguishing senescence traits. Aging 2013, 5, 98–99. [Google Scholar] [PubMed]
- Ross, A.L.; Sanchez, M.I.; Grichnik, J.M. Nevus senescence. ISRN Dermatol. 2011, 2011, 642157. [Google Scholar] [CrossRef] [PubMed]
- Aitken, J.; Welch, J.; Duffy, D.; Milligan, A.; Green, A.; Martin, N.; Hayward, N. CDKN2A variants in a population-based sample of queensland families with melanoma. J. Natl. Cancer Inst. 1999, 91, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Shattuck-Eidens, D.; Eeles, R.; Liu, Q.; Gruis, N.A.; Ding, W.; Hussey, C.; Tran, T.; Miki, Y.; Weaver-Feldhaus, J.; et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet. 1994, 8, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Cachia, A.R.; Indsto, J.O.; McLaren, K.M.; Mann, G.J.; Arends, M.J. CDKN2A mutation and deletion status in thin and thick primary melanoma. Clin. Cancer Res. 2000, 6, 3511–3515. [Google Scholar] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, mmac1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated AKT3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; de Vries, T.J.; Morandini, R.; Ghanem, G.G.; Ruiter, D.J.; van Muijen, G.N. E-cadherin expression in human melanoma. Melanoma Res. 1996, 6, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.Y.; Wheelock, M.J.; Johnson, K.R.; Herlyn, M. Shifts in cadherin profiles between human normal melanocytes and melanomas. J. Investig. Dermatol. Symp. Proc. 1996, 1, 188–194. [Google Scholar] [PubMed]
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. Beta-catenin-induced melanoma growth requires the downstream target microphthalmia-associated transcription factor. J. Cell Biol. 2002, 158, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the β-catenin/lef-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Moellmann, G.; Cheng, E.; Alvarez-Franco, M.; Wagner, S.; Sassone-Corsi, P.; Halaban, R. Identification of p90rsk as the probable creb-ser133 kinase in human melanocytes. Cell Growth Differ. 1995, 6, 291–302. [Google Scholar] [PubMed]
- Narita, N.; Tanemura, A.; Murali, R.; Scolyer, R.A.; Huang, S.; Arigami, T.; Yanagita, S.; Chong, K.K.; Thompson, J.F.; Morton, D.L.; et al. Functional ret G691s polymorphism in cutaneous malignant melanoma. Oncogene 2009, 28, 3058–3068. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Sanz-Moreno, V. Emerging molecular targets in melanoma invasion and metastasis. Pigment Cell Melanoma Res. 2013, 26, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Sharma, A.; Peng, H.H.; Robertson, G.; Dong, C. Targeting mutant (V600E) B-raf in melanoma interrupts immunoediting of leukocyte functions and melanoma extravasation. Cancer Res. 2007, 67, 5814–5820. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.S.; Madhunapantula, S.V.; Robertson, G.P.; Drabick, J.J. Current and future trials of targeted therapies in cutaneous melanoma. Adv. Exp. Med. Biol. 2013, 779, 223–255. [Google Scholar] [PubMed]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Hocker, T.; Tsao, H. Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Hum. Mutat. 2007, 28, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Giehl, K. Oncogenic ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, J.; Yuen, N.K.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A.; Demetri, G.D.; Widlund, H.R.; Fisher, D.E.; Hodi, F.S. Imatinib targeting of kit-mutant oncoprotein in melanoma. Clin. Cancer Res. 2008, 14, 7726–7732. [Google Scholar] [CrossRef] [PubMed]
- Kyrgidis, A.; Tzellos, T.G.; Triaridis, S. Melanoma: Stem cells, sun exposure and hallmarks for carcinogenesis, molecular concepts and future clinical implications. J. Carcinog. 2010, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic significance of activated akt expression in melanoma: A clinicopathologic study of 292 cases. J. Clin. Oncol. 2005, 23, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of bim expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Stemke-Hale, K.; Lin, E.; Tellez, C.; Deng, W.; Gopal, Y.N.; Woodman, S.E.; Calderone, T.C.; Ju, Z.; Lazar, A.J.; et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin. Cancer Res. 2009, 15, 7538–7546. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.K.; Lazar, A.J.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J. Investig. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. BRAF(V600E) cooperates with PTEN loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. WNT/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Weeraratna, A.T. Hear the WNT ROR: How melanoma cells adjust to changes in wnt. Pigment Cell Melanoma Res. 2009, 22, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Julius, M.A.; Giarre, M.; Zheng, Z.; Brown, A.M.; Kitajewski, J. Transformation by wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 1349–1358. [Google Scholar]
- Webster, M.R.; Weeraratna, A.T. A WNT-ER migration: The confusing role of beta-catenin in melanoma metastasis. Sci. Signal. 2013, 6, pe11. [Google Scholar] [CrossRef] [PubMed]
- Rimm, D.L.; Caca, K.; Hu, G.; Harrison, F.B.; Fearon, E.R. Frequent nuclear/cytoplasmic localization of β-catenin without exon 3 mutations in malignant melanoma. Am. J. Pathol. 1999, 154, 325–329. [Google Scholar] [CrossRef]
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. β-catenin induces immortalization of melanocytes by suppressing p16INK4A expression and cooperates with NRAS in melanoma development. Genes Dev. 2007, 21, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Straume, O.; Puntervoll, H.E.; Kalvenes, M.B.; Akslen, L.A. Importance of p-cadherin, β-catenin, and WNT5A/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 8606–8614. [Google Scholar] [CrossRef] [PubMed]
- Kageshita, T.; Hamby, C.V.; Ishihara, T.; Matsumoto, K.; Saida, T.; Ono, T. Loss of β-catenin expression associated with disease progression in malignant melanoma. Br. J. Dermatol. 2001, 145, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Toda, S.; Fujimoto, M.; Ohtsuki, M.; Byers, H.R.; Etoh, T.; Nakagawa, H. Constitutive activation of wnt/beta-catenin signaling pathway in migration-active melanoma cells: Role of lef-1 in melanoma with increased metastatic potential. Biochem. Biophys. Res. Commun. 2001, 288, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zuidervaart, W.; Pavey, S.; van Nieuwpoort, F.A.; Packer, L.; Out, C.; Maat, W.; Jager, M.J.; Gruis, N.A.; Hayward, N.K. Expression of wnt5a and its downstream effector β-catenin in uveal melanoma. Melanoma Res. 2007, 17, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Ross, K.; Huber, W.E.; Nishimura, E.K.; Golub, T.R.; Fisher, D.E. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by mitf. Cancer Cell 2004, 6, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. MITF regulation of DIA1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.E.; Levy, C. The three m’s: Melanoma, microphthalmia-associated transcription factor and microrna. Pigment Cell Melanoma Res. 2011, 24, 1088–1106. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Rana, S.; Paterson, H.; Pickersgill, H.; Brummelkamp, T.; Marais, R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor mitf. PLoS ONE 2008, 3, e2734. [Google Scholar] [CrossRef] [PubMed]
- Du, S.J.; Purcell, S.M.; Christian, J.L.; McGrew, L.L.; Moon, R.T. Identification of distinct classes and functional domains of wnts through expression of wild-type and chimeric proteins in xenopus embryos. Mol. Cell. Biol. 1995, 15, 2625–2634. [Google Scholar] [PubMed]
- Seifert, J.R.; Mlodzik, M. Frizzled/pcp signalling: A conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 2007, 8, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.P.; Bradley, A.; McMahon, A.P.; Jones, S. A wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 1999, 126, 1211–1223. [Google Scholar] [PubMed]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. WNT5A signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Eichstetter, I.; Danielson, K.G. Aberrant expression of the growth factor WNT5A in human malignancy. Cancer Res. 1995, 55, 3495–3499. [Google Scholar] [PubMed]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Da Forno, P.D.; Pringle, J.H.; Hutchinson, P.; Osborn, J.; Huang, Q.; Potter, L.; Hancox, R.A.; Fletcher, A.; Saldanha, G.S. WNT5A expression increases during melanoma progression and correlates with outcome. Clin. Cancer Res. 2008, 14, 5825–5832. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Milovanovic, T.; Barr, R.J.; Truong, T.; Holcombe, R.F. Wnt ligand expression in malignant melanoma: Pilot study indicating correlation with histopathological features. Mol. Pathol. 2003, 56, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Talantov, D.; Mazumder, A.; Yu, J.X.; Briggs, T.; Jiang, Y.; Backus, J.; Atkins, D.; Wang, Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 2005, 11, 7234–7242. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The WNT5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Fiori, J.L.; Xu, M.; Carter, A.D.; Frank, B.P.; Camilli, T.C.; French, A.D.; Dissanayake, S.K.; Indig, F.E.; Bernier, M.; et al. The orphan tyrosine kinase receptor, ROR2, mediates WNT5A signaling in metastatic melanoma. Oncogene 2010, 29, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, A.H.; Yoo, J.H.; Clancy, J.; Sorensen, L.K.; Sedgwick, A.; Tong, Z.; Ostanin, K.; Rogers, A.; Grossmann, K.F.; Tripp, S.R.; et al. The small gtpase ARF6 stimulates β-catenin transcriptional activity during wnt5a-mediated melanoma invasion and metastasis. Sci. Signal. 2013, 6, ra14. [Google Scholar] [CrossRef] [PubMed]
- Eichhoff, O.M.; Zipser, M.C.; Xu, M.; Weeraratna, A.T.; Mihic, D.; Dummer, R.; Hoek, K.S. The immunohistochemistry of invasive and proliferative phenotype switching in melanoma: A case report. Melanoma Res. 2010, 20, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Eichhoff, O.M.; Weeraratna, A.; Zipser, M.C.; Denat, L.; Widmer, D.S.; Xu, M.; Kriegl, L.; Kirchner, T.; Larue, L.; Dummer, R.; et al. Differential lef1 and tcf4 expression is involved in melanoma cell phenotype switching. Pigment Cell Melanoma Res. 2011, 24, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Jenei, V.; Sherwood, V.; Howlin, J.; Linnskog, R.; Safholm, A.; Axelsson, L.; Andersson, T. A T-butyloxycarbonyl-modified WNT5A-derived hexapeptide functions as a potent antagonist of wnt5a-dependent melanoma cell invasion. Proc. Natl. Acad. Sci. USA 2009, 106, 19473–19478. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Chaurasiya, S.K.; Ekstrom, E.J.; Guilmain, W.; Liu, Q.; Koeck, T.; Brown, K.; Hansson, K.; Agnarsdottir, M.; Bergqvist, M.; et al. WNT5A-mediated beta-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis 2014, 35, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Linnskog, R.; Jonsson, G.; Axelsson, L.; Prasad, C.P.; Andersson, T. Interleukin-6 drives melanoma cell motility through p38α-MAPK-dependent up-regulation of WNT5A expression. Mol. Oncol. 2014, 8, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Karasarides, M.; Hayward, R.; Marais, R. Tumor necrosis factor-α blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res. 2007, 67, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in braf-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with mek inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Halait, H.; Demartin, K.; Shah, S.; Soviero, S.; Langland, R.; Cheng, S.; Hillman, G.; Wu, L.; Lawrence, H.J. Analytical performance of a real-time pcr-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn. Mol. Pathol. 2012, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Rosen, N. Towards a unified model of raf inhibitor resistance. Cancer Discov. 2014, 4, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Todd, J.R.; Gowrishankar, K.; Mijatov, B.; Pupo, G.M.; Fung, C.; Snoyman, S.; Hersey, P.; Long, G.V.; Kefford, R.F.; et al. Differential activity of mek and erk inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 2014, 8, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a raf kinase switch in melanoma can be overcome by cotargeting mek and igf-1r/pi3k. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or NRAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Marsh Durban, V.; Deuker, M.M.; Bosenberg, M.W.; Phillips, W.; McMahon, M. Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J. Clin. Investig. 2013, 123, 5104–5118. [Google Scholar] [CrossRef] [PubMed]
- Deuker, M.M.; Marsh Durban, V.; Phillips, W.A.; McMahon, M. Π3′-kinase inhibition forestalls the onset of mek1/2 inhibitor resistance in BRAF-mutated melanoma. Cancer Discov. 2015, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lassen, A.; Atefi, M.; Robert, L.; Wong, D.J.; Cerniglia, M.; Comin-Anduix, B.; Ribas, A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 2014, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or src family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Erin Chen, Y.; Kumar, R.; Taylor, M.; Jenny Njauw, C.N.; Miao, B.; Frederick, D.T.; Wargo, J.A.; Flaherty, K.T.; Jonsson, G.; et al. Mitf modulates therapeutic resistance through EGFR signaling. J. Investig. Dermatol. 2015, 123, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Gong, K.W.; Dering, J.; Tseng, Y.; Ginther, C.; Pauletti, G.; Glaspy, J.A.; Essner, R.; Bollag, G.; Hirth, P.; et al. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with plx4032 in malignant melanoma. Neoplasia 2010, 12, 637–649. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ror1 and ror2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.T.; Chen, H.W.; Haydu, L.; Toroni, R.A.; et al. Wnt5a enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Nambotin, S.B.; Lefrancois, L.; Sainsily, X.; Berthillon, P.; Kim, M.; Wands, J.R.; Chevallier, M.; Jalinot, P.; Scoazec, J.Y.; Trepo, C.; et al. Pharmacological inhibition of frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J. Hepatol. 2011, 54, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chua, M.S.; Grepper, S.; So, S.K. Soluble frizzled-7 receptor inhibits WNT signaling and sensitizes hepatocellular carcinoma cells towards doxorubicin. Mol. Cancer 2011, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An antagonist of dishevelled protein-protein interaction suppresses β-catenin-dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Biechele, T.L.; Kulikauskas, R.M.; Toroni, R.A.; Lucero, O.M.; Swift, R.D.; James, R.G.; Robin, N.C.; Dawson, D.W.; Moon, R.T.; Chien, A.J. WNT/β-catenin signaling and axin1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci. Signal. 2012, 5, ra3. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.R.; Xu, M.; Kinzler, K.A.; Kaur, A.; Appleton, J.; O’Connell, M.P.; Marchbank, K.; Valiga, A.; Dang, V.M.; Perego, M.; et al. WNT5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D.; et al. WNT5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef] [PubMed]
- Von Felbert, V.; Cordoba, F.; Weissenberger, J.; Vallan, C.; Kato, M.; Nakashima, I.; Braathen, L.R.; Weis, J. Interleukin-6 gene ablation in a transgenic mouse model of malignant skin melanoma. Am. J. Pathol. 2005, 166, 831–841. [Google Scholar] [CrossRef]
- Hoejberg, L.; Bastholt, L.; Johansen, J.S.; Christensen, I.J.; Gehl, J.; Schmidt, H. Serum interleukin-6 as a prognostic biomarker in patients with metastatic melanoma. Melanoma Res. 2012, 22, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Levin, R.S.; Gordan, J.D.; Oses-Prieto, J.A.; Webber, J.T.; Salt, M.; Hann, B.; Burlingame, A.L.; McCormick, F.; Bandyopadhyay, S.; et al. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 2014, 8, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Kibler, A.; Phelps, D.A.; Manap, S.; Xiao, L.; Lin, J.; Capper, D.; Oswald, D.; Geier, B.; DeWire, M.; et al. Development, characterization, and reversal of acquired resistance to the mek1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin. Cancer Res. 2013, 19, 6716–6729. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. Raf inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired b-raf inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to raf inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. Mek1 mutations confer resistance to mek and b-raf inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to raf inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Koya, R.C.; Nazarian, R.; Pupo, G.M.; Bacchiocchi, A.; Dahlman, K.B.; Chmielowski, B.; Sosman, J.A.; et al. Preexisting mek1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2012, 2, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. Map kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates raf dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF (V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, C.P.; Mohapatra, P.; Andersson, T. Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers 2015, 7, 1900-1924. https://doi.org/10.3390/cancers7030868
Prasad CP, Mohapatra P, Andersson T. Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers. 2015; 7(3):1900-1924. https://doi.org/10.3390/cancers7030868
Chicago/Turabian StylePrasad, Chandra Prakash, Purusottam Mohapatra, and Tommy Andersson. 2015. "Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer?" Cancers 7, no. 3: 1900-1924. https://doi.org/10.3390/cancers7030868
APA StylePrasad, C. P., Mohapatra, P., & Andersson, T. (2015). Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers, 7(3), 1900-1924. https://doi.org/10.3390/cancers7030868