
The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer

Abstract
:1. Introduction
2. Adiposity, Inflammation and Type 2 Diabetes

{kind=link}
{kind=link}
| Functions | Macrophage Phenotype | ||
|---|---|---|---|
| M1 | M2 | ||
| Phenotypic induction | Proinflammatory | Anti-inflammatory Anti-insulin resistance IL-10 (as induced by adiponectin), plus IL-4 and 3 | |
| Pro-insulin resistance | |||
| INF-γ with LPS | |||
| M2a “alternatively activated” | M2b “regulatory” | ||
| Cytokines | TNF-α | IL-4 | IL-10 |
| IL-6 | |||
| IL-12 | |||
| IL-1β | |||
| Chemokine | MCP-1 * | MCP-1 * | |
| Bioactivity | Activates NF-κB | Suppresses NF-κB; Pro-collagen synthesis; Anti-inflammation tissue repair | |
3. Obesity, Type 2 Diabetes and Breast Cancer
3.1. Obesity
3.2. Hyperinsulinemia
3.3. Type 2 Diabetes
| Reference (No.) | T1 | T2 | T3 + T4 | Positive Lymph Nodes | BMI | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| ND % | D % | ND % | D % | ND % | D % | ND % | D % | ND | D | |
| [73] | 67.8 | 62.8 | 26.4 | 30.9 | 4.8 | 5.3f | 22.1 | 24.8f | not given | |
| [74] | 57.9 | 48.0 | 35.0 | 41.2 | 7.1 | 10.8 ** | 34.9 | 41.1 * | 25.9 ± 4.7 | 28.8 ± 5.6f |
| [79] | 55.5 | 33.8 | 37.3 | 55.4 | 7.2 | 10.8 * | 24.0 | 33.0 | 26.9 ± 4.1 | 29.7 ± 4.5f |
| [80] | 34.3 | 25.9 | 50.3 | 55.9 | 15.4 | 18.2 | 43.4 | 56.6f | 26.9 ± 4.0 | 27.1 ± 3.8 |
| [81] | 18.0 | 10.5 | 60.5 | 62.9 | 21.5 | 26.7 | 53.0 | 66.0 * | not given | |
| [82] | 35.5 | 28.5 | 52.2 | 55.2 | 12.3 | 16.3f | 43.3 | 47.5 * | 28.1% | 47.5%f |
3.4. Obesity-Insulin Resistance Interaction and Breast Cancer
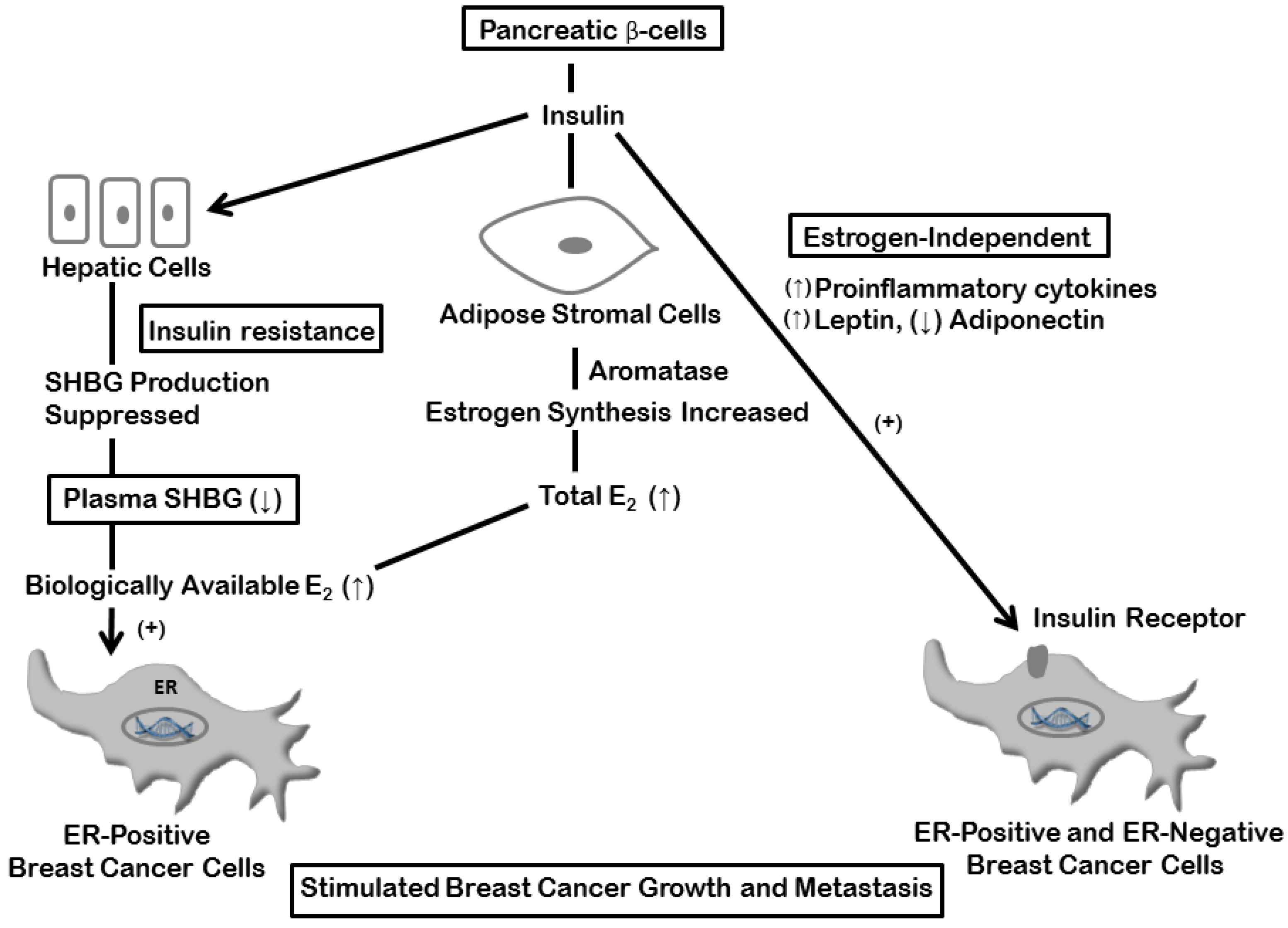
4. Estrogen and Insulin Mechanisms

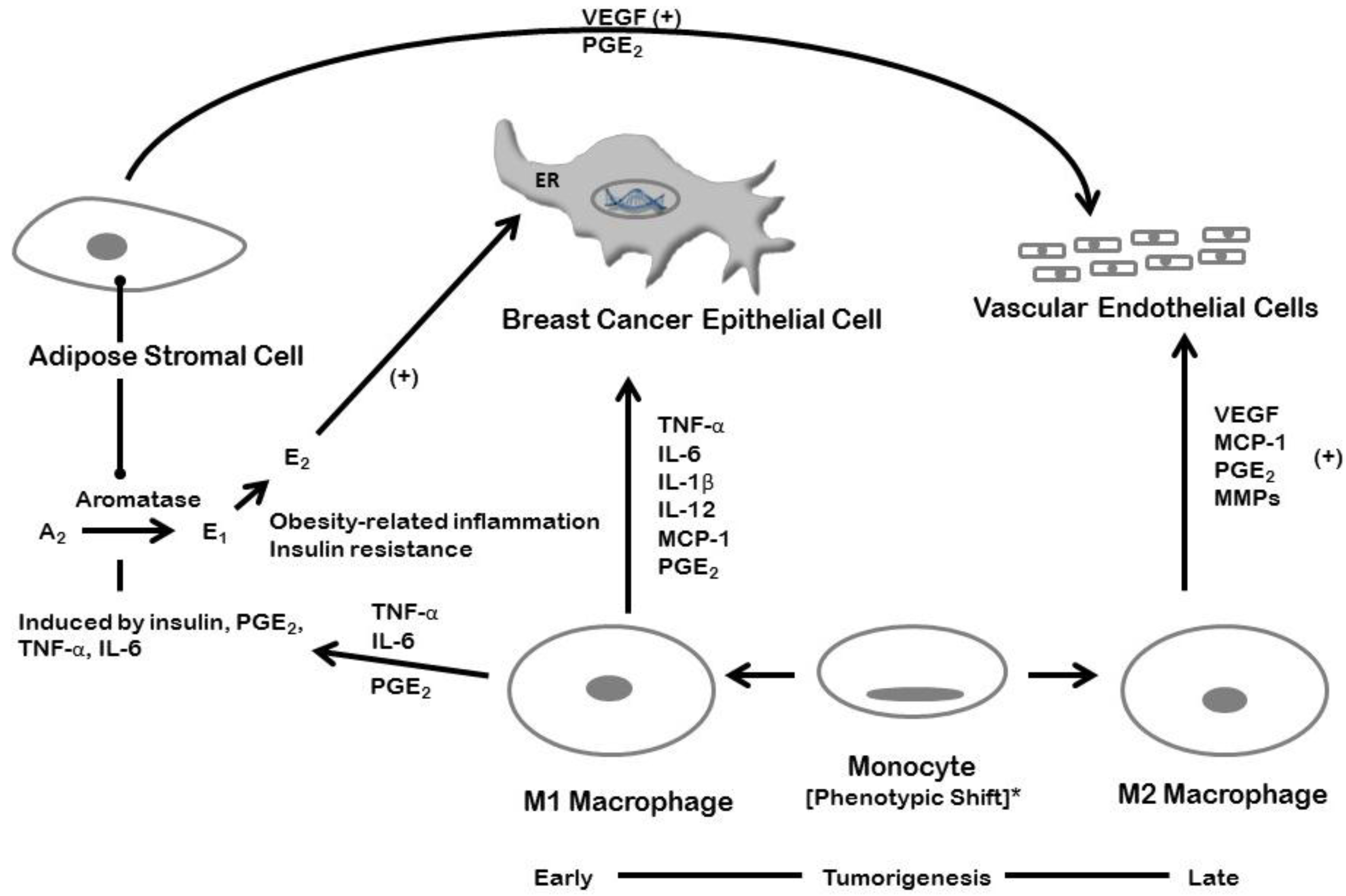
5. Inflammation and Breast Cancer: Estrogen-dependent Mechanisms

6. Inflammation and Breast Cancer: Estrogen-Independent Mechanisms
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2014. [Google Scholar]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- De Waard, F.; Baanders-van Halewijin, E.A.; Huizinga, J. The bimodal age distribution of patients with mammary carcinoma. Evidence for the existence of 2 types of human breast cancer. Cancer 1964, 17, 141–151. [Google Scholar] [PubMed]
- De Waard, F.; Baanders-van Halewijn, E.A. A prospective study in general practice on breast-cancer risk in postmenopausal women. Int. J. Cancer. 1974, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999–2010. J. Am. Med. Assoc. 2012, 307, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Cowie, C.C.; Rust, K.F.; Ford, E.S.; Eberhardt, M.S.; Byrd-Holt, D.D.; Li, C.; Williams, D.E.; Gregg, E.W.; Bainbridge, K.E.; Saydah, S.H.; et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care. 2009, 32, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Coldlitz, G.A.; Willet, W.C.; Stampfer, M.J.; Manson, J.E.; Hennekens, C.H.; Arky, R.A.; Speizer, F.E. Weight as a risk factor for clinical diabetes in women. Am. J. Epidemiol. 1990, 132, 501–513. [Google Scholar]
- Cali, A.M.; Caprio, S. Prediabetes and type 2 diabetes in youth: An emerging epidemic disease? Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Review series Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Onat, A.; Avci, G.S.; Barlan, M.M.; Uyarel, H.; Uzunlar, B.; Sansoy, V. Measures of abdominal obesity assessed for visceral adiposity and relation to coronary risk. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.S.; Ruderman, M.B. Adipose tissue inflammation and insulin resistance: All obese humans are not created equal. Biochem. J. 2010, 430, E1–E4. [Google Scholar] [CrossRef] [PubMed]
- Goran, M.I.; Alderete, T.L. Targeting adipose tissue inflammation to treat the underlying basis of the metabolic complications of obesity. Nestle Nutr. Inst. Workshop Ser. 2012, 73, 49–60. [Google Scholar] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W.; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Barbarroja, N.; Lopez-Pedera, R.; Mayas, M.D.; Garcia-Fuentes, E.; Garrido-Sanchez, L.; Macias-Gonzalez, M.; El Bekay, R.; Vidal-Puig, A.; Tinahones, F.J. The obese healthy paradox: Is inflammation the answer? Biochem. J. 2010, 430, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Barbarroja, N.; Lopez-Pedrera, C.; Garrido-Sanchez, L.; Mayas, M.D.; Oliva-Olivera, W.; Bernal-Lopez, M.R.; El Bekay, R.; Tinahones, F.J. Progression from high insulin resistance to type 2 diabetes does not entail additional visceral adipose tissue inflammation. PLOS ONE 2012, 7. [Google Scholar] [CrossRef]
- Chen, A.; Mumick, S.; Zhang, C.; Lamb, J.; Dai, H.; Weingarth, D.; Mudgett, J.; Chen, H.; MacNeil, D.J.; Reitman, M.L.; et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 2005, 13, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Bruce, C.R.; Sacchetti, M.; Anderson, M.J.; Olsen, D.B.; Saltin, B.; Hawley, J.A.; Febbraio, M. Interleukin-6 and tumor necrosis factor-alpha are not increased in patients with Type 2 diabetes: Evidence that plasma interleukin-6 is related to fat mass and not insulin responsiveness. Diabetologia 2004, 47, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Bal, Y.; Adas, M.; Helvaci, A. Evaluation of the relationship between insulin resistance and plasma tumor necrosis factor-alpha, interleukin-6 and C-reactive protein levels in obese women. Bratisl. Lek. Listy 2010, 111, 200–204. [Google Scholar] [PubMed]
- Lê, K.A.; Mahurkar, S.; Alderete, T.L.; Hasson, R.E.; Adam, T.C.; Kim, J.S.; Beale, E.; Xie, C.; Greenberg, A.S.; Allayee, H.; et al. Subcutaneous adipose tissue macrophage infiltration is associated with hepatic and visceral fat deposition, hyperinsulinemia, and stimulation of NF-kB stress pathway. Diabetes 2011, 60, 2802–2809. [Google Scholar]
- Bremer, A.A.; Devaraj, J.; Afify, A.; Jilal, I. Adipose tissue dysregulation in patients with metabolic syndrome. J. Clin. Endocrinol. Meab. 2011, 96, E1782–E1788. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Chae, J.S.; Paik, J.K.; Seo, H.S.; Jang, Y.; Cavailon, J.M.; Lee, J.H. Effects of aging and menopause on serum interleukin-6 levels and peripheral blood mononuclear cell cytokine production in healthy nonobese women. Age 2012, 2, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp. Cell Res. 2011, 317, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Bao, S.; Bolin, E.R.; Burris, D.L.; Xu, X.; Sun, Q.; Killilea, D.W.; Shen, Q.; Ziouzenkova, O.; Belury, M.; et al. Zinc deficiency augments leptin production and exacerbates macrophage infiltration into adipose tissue in mice fed a high-fat diet. J. Nutr. 2013, 143, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, L.K.; Campbell, L.V. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr. Pharm. Des. 2008, 14, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Inoguchi, T.; Batchuluun, B.; Sugiyama, N.; Kobayashi, K.; Sonoda, N.; Takayanagi, R. CTLA-4Ig immunotherapy of obesity-induced insulin resistance by manipulation of macrophage polarization in adipose tissues. Biochem. Biophys. Res. Commun. 2013, 438, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Dendale, P.; Beelen, M.; Jonkers, R.M.; Mullens, A.; Corluy, L.; Meeusen, R.; van Loon, L.J.C. Plasma adipokine and inflammatory marker concentrations are altered in obese, as opposed to non-obese, type 2 diabetes patients. Eur. J. Appl. Physiol. 2010, 109, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Engeli, S.; Feldpausch, M.; Gorzelniak, K.; Hartwig, F.; Heintze, U.; Janke, J.; Möhlig, M.; Pfeiffer, A.F.; Luft, F.C.; Sharma, A.M. Association between adiponectin and mediators of inflammation in obese women. Diabetes 2003, 52, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gocke, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Summer, R.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Mohamed-Ali, V.; Horakova, O.; Rossmeisl, M.; Hosseinzadeh-Attar, M.J.; Hensler, M.; Ruzickova, J.; Kopecky, J. Polyunsaturated fatty acids of marine origin induce adiponectin in mice fed a high-fat diet. Diabetologia 2006, 49, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Micallef, M.; Munro, I.; Phang, M.; Garg, M. Plasma n-3 Polyunsaturated Fatty Acids are negatively associated with obesity. Br. J. Nutr. 2009, 102, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Shouzu, A.; Omoto, S.; Inami, N.; Ueba, T.; Urase, F.; Maeda, Y. Effects of eicosapentaenoic acid on endothelial cell-derived microparticles, angiopoietins and adiponectin in patients with type 2 diabetes. J. Atheroscler. Thromb. 2009, 16, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer 2007, 14, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. Type 2 diabetes and obesity metabolic interactions: Common factors for breast cancer risk and novel approaches to prevention and therapy. Curr. Diabetes Rev. 2012, 8, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar]
- Ndumele, C.E.; Nasir, K.; Conceiçao, R.D.; Carvalho, J.A.M.; Blumenthal, R.S.; Santos, R.D. Hepatic steatosis, obesity, and the metabolic syndrome are independently and additively associated with increased systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLOS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 Diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Machann, J.; Schick, F.; Thamer, C.; Rittig, K.; Balletshofer, B.; Machicao, F.; Fritsche, A.; Häring, H.U. Identification and characterization of metabolically benign obesity in humans. Arch. Intern. Med. 2008, 168, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; Mcginn, A.P. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering. Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Perry, I.J. Does inflammation determine metabolic health status in obese and nonobese adults? J. Clin. Endocrinol. Metab. 2013, 98, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Conus, F.; Rabasa-Lhoret, R.; Peronnet, F. Characteristics of metabolically obese normal-weight (MONW) subjects. Appl. Physiol. Nutr. Metab. 2007, 32, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wildman, R.P.; Kaplan, R.; Manson, J.E.; Rajkovic, A.; Connelly, S.A.; Mackey, R.H.; Tinker, L.F.; Curb, J.D.; Eaton, C.B.; Wassertheil-Smoller, S. Body size phenotypes and inflammation in the Women’s Health Initiative Observational Study. Obesity 2011, 19, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. Biochemical and molecular mechanisms for the association between obesity, chronic inflammation, and breast cancer. Biofactors 2013, 40, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Manson, J.E.; Rosner, B.; Speizer, F.E.; Hankinson, S.E. Waist circumference, waist:hip ratio, and risk of breast cancer in the Nurses’ Health Study. Am. J. Epidemiol. 1999, 150, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Barnett, C.; Vogt, K.N.; Li, T.; Stone, J.; Boyd, N.F. A meta-analysis of published literature on waist-to-hip ratio and risk of breast cancer. Nutr. Cancer 2002, 44, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-Analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niraula, S.; Ocana, A.; Ennis, M.; Goodwin, P.J. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: A meta-analysis. Breast Cancer Res. Treat. 2012, 134, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Sestak, I.; Distler, W.; Forbes, J.F.; Dowsett, M.; Howell, A.; Cuzick, J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. J. Clin. Oncol. 2010, 28, 3411–3415. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, G.; Stöger, H.; Dubsky, P.; Mlineritsch, B.; Singer, C.; Balic, M.; Fitzal, F.; Moik, M.; Kwasny, W.; Selim, U.; et al. Efficacy of tamoxifen ± aminoglutethimide in normal weight and overweight postmenopausal patients with hormone receptor-positive breast cancer: An analysis of 1509 patients of the ABCSG-06 trial. Br. J. Cancer 2013, 108, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Autier, P.; Koechlin, A.; Boniol, M.; Mullie, P.; Bolli, G.; Rosenstock, J.; Boyle, P. Serum insulin and C-peptide concentration and breast cancer: A meta-analysis. Cancer Causes Control. 2013, 24, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Lundin, E.; Manjer, J.; Rinaldi, S.; Biessy, C.; Söderberg, S.; Lenner, P.; Janzon, L.; Riboli, E.; Berglund, G.; et al. Prospective study of IGF-I, IGF-binding proteins, and breast cancer risk, in northern and southern Sweden. Cancer Causes Control 2002, 13, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Mink, P.J.; Shahar, E.; Rosamond, W.D.; Alberg, A.J.; Folsom, A.R. Serum insulin and glucose levels and breast cancer incidence: The atherosclerosis risk in communities study. Am. J. Epidemiol. 2002, 156, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Andersen, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Kabat, G.C.; Kim, M.; Caan, B.J.; Chlebowski, R.T.; Gunter, M.J.; Ho, G.Y.F.; Rodriguez, B.L.; Shikany, J.M.; Strickler, H.D.; Vitolins, M.Z.; et al. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int. J. Cancer 2009, 125, 2704–2710. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Suehiro, T.; Nakamura, T.; Kumon, Y.; Hashimoto, K. Clinical significance of the insulin resistance index as assessed by homeostasis model assessment. Endocr. J. 2001, 48, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Taylor, S.K.; Hood, N. Insulin- and obesity-related variables in early-stage breast cancer: Correlations and time course of prognostic associations. J. Clin. Oncol. 2012, 30, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Borugian, M.J.; Sheps, S.B.; Kim-Sing, C.; van Patten, C.; Potter, J.D.; Dunn, B.; Gallagher, R.P.; Hislop, T.G. Insulin, macronutrient intake, and physical activity: Are potential indicators of insulin resistance associated with mortality from breast cancer? Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1163–1172. [Google Scholar] [PubMed]
- Rose, D.P.; Vona-Davis, L. Influence of obesity on breast cancer receptor status and prognosis. Expert Rev. Anticancer Ther. 2009, 9, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.L.; Boniol, M.; Zheng, T.; Zhang, Y.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, K.M.J.; Arends, L.R.; Hansen, B.E.; Leeflang, S.; Ruiter, R.; van Eijck, C.H.J. Systematic review and meta-analysis of the association between diabetes mellitus and incidence and mortality in breast and colorectal cancer. Br. J. Surg. 2013, 100, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Peairs, K.S.; Barone, B.B.; Snyder, C.F.; Yeh, H.C.; Stein, K.B.; Derr, R.L.; Brancati, F.L.; Wolff, A.C. Diabetes mellitus and breast cancer outcomes: A systematic review and meta-analysis. J. Clin. Oncol. 2011, 29, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.T.; Rastogi, A.; Dmitrienko, A.; Johnson, K.D. A comprehensive prognostic index to predict survival based on multiple comorbidities: A focus on breast cancer. Med. Care 1999, 37, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Srokowski, T.P.; Fang, S.; Hortobagyi, G.N.; Giordano, S.H. Impact of diabetes mellitus on complications and outcomes of adjuvant chemotherapy in older patients with breast cancer. J. Clin. Oncol. 2009, 27, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Schrauder, M.G.; Fasching, P.A.; Häberle, L.; Lux, M.P.; Rauh, C.; Hein, A.; Bayer, C.M.; Heusinger, K.; Hartmann, A.; Strehl, J.D.; et al. Diabetes and prognosis in a breast cancer cohort. J. Cancer Res. Clin. Oncol. 2011, 137, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Kim, E.S.; Dong, W.; Feng, L.; Hortobagyi, G.N.; Giordano, S.H. Obesity, diabetes, and survival outcomes in a large cohort of early-stage breast cancer patients. Ann. Oncol. 2013, 24, 2506–5214. [Google Scholar] [CrossRef] [PubMed]
- Druesne-Pecollo, N.; Touvier, M.; Barrandon, E.; Chan, D.S.; Norat, T.; Zelek, L.; Hercberg, S.; Latino-Martel, P. Excess body weight and second primary cancer risk after breast cancer: A systematic review and meta-analysis of prospective studies. Breast Cancer Res. Treat. 2012, 135, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Daling, J.R.; Tang, M.T.; Malone, K.E. Relationship between diabetes and risk of second primary contralateral breast cancer. Breast Cancer Res. Treat. 2011, 125, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Sadetzki, S.; Gluck, I.; Oberman, B.; Ben-David, M.; Papa, M.Z.; Catane, R.; Kaufman, B. Association between diabetes mellitus and adverse characteristics of breast cancer at presentation. Eur. J. Cancer 2006, 42, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Li, J.; Wang, L.; Zhang, Y.; Wang, C.; Hu, M.; Ma, B.; Wang, G.; Sun, S. Type 2 diabetes mellitus and characteristics of breast cancer in China. Asian Pac. J. Cancer Prev. 2010, 11, 933–937. [Google Scholar] [PubMed]
- Li, Z.; Luo, Y.; Gong, Y.; Liu, Y.; Qiu, W.; Tu, J. Clinical features and molecular phenotypes of breast cancer in patients with type-2 diabetes mellitus. Asian Pac. J. Cancer Prev. 2011, 12, 2183–2188. [Google Scholar] [PubMed]
- Hou, G.; Zhang, S.; Zhang, X.; Wang, P.; Hao, X.; Zhang, J. Clinical pathological characteristics and prognostic analysis of 1,013 breast cancer patients with diabetes. Breast Cancer Res. Treat. 2013, 137, 807–816. [Google Scholar] [CrossRef] [PubMed]
- De Azambuja, E.; Cardoso, F.; de Castro, G.; Colozza, M.; Mano, M.S.; Durbecq, V.; Sotiriou, C.; Larsimont, D.; Piccart-Gebhart, M.J.; Paesmans, M. Ki-67 as prognostic marker in early breast cancer: A meta-analysis of published studies involving 12,155 patients. Br. J. Cancer 2007, 96, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Kameni, A.; Anderson, M.L.; White, E.; Taplin, S.H.; Porter, P.; Ballard-Barbash, R.; Malone, K.; Buist, D.S. Body mass index, tumor characteristics, and prognosis following diagnosis of early-stage breast cancer in a mammographically screened population. Cancer Causes Control 2013, 24, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Moulder-Thompson, S.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.B.; Solomon, C.G.; Hu, F.B.; Rosner, B.A.; Hankinson, S.E.; Colditz, G.A.; Manson, J.E. Type 2 diabetes and subsequent incidence of breast cancer in the nurses’ health study. Diabetes Care 2003, 26, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Amadou, A.; Ferrari, P.; Muwonge, R.; Moskal, A.; Biessy, C.; Romieu, I.; Hainaut, P. Overweight, obesity and risk of premenopausal breast cancer according to ethnicity: A systematic review and dose-response meta-analysis. Obes. Rev. 2013, 14, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Yu, M.C.; Tseng, C.C.; Stanczyk, F.Z.; Pike, M.C. Diabetes and risk of breast cancer in Asian-American women. Carcinogenesis 2007, 28, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Razak, F.; Anand, S.S.; Shannon, H.; Vuksan, V.; Davis, B.; Jacobs, R.; Teo, K.K.; McQueen, M.; Yusuf, S. Defining obesity cut points in a multiethnic population. Circulation 2007, 115, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G. Endogenous Sex Hormones and Breast Cancer in Postmenopausal Women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar] [PubMed]
- Rinaldi, S.; Key, T.J.; Peeters, P.H.M.; Lahmann, P.H.; Lukanova, A.; Dossus, L.; Biessy, C.; Vineis, P.; Sacerdote, C.; Berrino, F.; et al. Anthropometric measures, endogenous sex steroids and breast cancer risk in postmenopausal women: A study within the EPIC cohort. Int. J. Cancer 2006, 118, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. Interaction between menopausal status and obesity in affecting breast cancer risk. Maturitas 2010, 66, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tekmal, R.R.; Kirma, N.; Gill, K.; Fowler, K. Aromatase overexpression and breast hyperplasia, an in vivo model-continued overexpression of aromatase is sufficient to maintain hyperplasia without circulating estrogens, and aromatase inhibitors abrogate these preneoplastic changes in mammary glands. Endocr. Relat. Cancer 1999, 6, 307–314. [Google Scholar] [CrossRef] [PubMed]
- McTernan, P.G.; Anwar, A.; Eggo, M.C.; Barnett, A.H.; Stewart, P.M.; Kumar, S. Gender differences in the regulation of P450 aromatase expression and activity in human adipose tissue. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 875–881. [Google Scholar] [PubMed]
- Ding, E.L.; Song, Y.; Manson, J.E.; Rifai, N.; Buring, J.E.; Liu, S. Plasma sex steroid hormones and risk of developing type 2 diabetes in women: A prospective study. Diabetologia 2007, 50, 2076–2084. [Google Scholar] [PubMed]
- Kalyani, R.R.; Franco, M.; Dobs, A.S.; Ouyang, P.; Vaidya, D.; Bertoni, A.; Gapstur, S.M.; Golden, S.H. The association of endogenous sex hormones, adiposity, and insulin resistance with incident diabetes in postmenopausal women. J. Clin. Endocrinol. Metab. 2009, 94, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Daka, B.; Jansson, P.A.; Rosen, T.; Larsson, C.A.; Rastam, L.; Lindblad, U. Inverse association between serum insulin and sex hormone binding globulin in a population survey in Sweden. Endocr. Connect. 2012, 2, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.H.; Dobs, A.S.; Vaidya, D.; Szklo, M.; Gapstur, S.; Kopp, P.; Liu, K.; Ouyang, P. Endogenous sex hormones and glucose tolerance status in postmenopausal women. J. Clin. Endocrinol. Metab. 2007, 92, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Hortobagyi, G.N. Prognostic molecular markers in early breast cancer. Breast Cancer Res. 2004, 6, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berstein, L.M.; Tsrylina, E.V.; Poroshina, T.E.; Levina, V.V.; Vasilyev, D.A.; Kovalenko, I.G.; Semiglazov, V.F. Genotoxic factors associated with the development of receptor-negative breast cancer: Potential role of the phenomenon of switching of estrogen effects. Exp. Oncol. 2006, 28, 64–69. [Google Scholar] [PubMed]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Glycemic load, glycemic index and breast cancer risk in a prospective cohort of Swedish women. Int. J. Cancer 2009, 125, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr. Relat. Cancer 2012, 19, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Casbas-Hernandez, P.; Bigelow, C.; Makowski, L.; Joseph Jerry, D.; Smith Schneider, S.; Troester, M.A. Normal breast tissue of obese women is enriched for macrophage markers and macrophage-associated gene expression. Breast Cancer Res. Treat. 2012, 131, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L.; et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Nichols, J.E.; Valdez, R.; Mendelson, C.R.; Simpson, E.R. Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol. Endocrinol. 1996, 11, 1350–1357. [Google Scholar]
- Singh, A.; Purohit, A.; Ghilchik, M.W.; Reed, M.J. The regulation of aromatase activity in breast fibroblasts: The role of interleukin-6 and prostaglandin E2. Endocr. Relat. Cancer 1999, 6, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Morris, P.G.; Zhou, X.K.; Morrow, M.; Du, B.; Giri, D.; Kopelovich, L.; Hudis, C.A.; Dannenberg, A.J. Increased levels of COX-2 and prostaglandin E 2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012, 2, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.H.; Li, J.W.P.; Strissel, K.J.; Obin, M.S.; Greenberg, A.S. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 2009, 150, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Weaver, A.; Pradhan, M.; Dai, Y.; Miller, L.D.; Lin, C.Y.; Stanculescu, A. Positive cross-talk between estrogen receptor and NF-κB in breast cancer. Cancer Res. 2009, 69, 8918–8925. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Rojo, F.; A’Hern, R.; Villena, N.; Salter, J.; Corominas, J.M.; Servitja, S.; Smith, I.E.; Rovira, A.; Reis-Filho, J.S.; et al. Nuclear NF-κB/p65 expression and response to neoadjuvant chemotherapy in breast cancer. J. Clin. Pathol. 2011, 62, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Choi, M.R.; Park, H.; Kim, M.; Hong, J.; Lee, J.Y.; Chun, H.; Lee, K.; Yoon Park, J. Dietary fat increases solid tumor growth and metastasis of 4T1 murine mammary carcinoma cells and mortality in obesity-resistant BALB/c mice. Breast Cancer Res. 2011, 13, R78. [Google Scholar] [CrossRef] [PubMed]
- Berryhill, G.E.; Gloviczki, J.M.; Trott, J.F.; Aimo, L.; Kraft, J.; Cardiff, R.D.; Paul, C.T.; Petrie, W.K.; Lock, A.L.; Hovey, R.C. Diet-induced metabolic change induces estrogen-independent allometric mammary growth. Proc. Natl. Acad. Sci. 2012, 109, 16294–16299. [Google Scholar] [CrossRef] [PubMed]
- Poirier, H.; Shapiro, J.S.; Kim, R.J.; Lazar, M.A. Nutritional supplementation with trans-10, cis-12-conjugated linoleic acid induces inflammation of white adipose tissue. Diabetes 2006, 55, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Cowen, S.; McLaughlin, S.; Hobbs, G.; Coad, J.; Martin, K.; Olfert, I.; Vona-Davis, L. High-fat, high-calorie diet enhances mammary carcinogenesis and local inflammation in MMTV-PyMT mouse model of breast cancer. Cancers 2015, 7, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Francés, D.E.; Motiño, O.; Agrá, N.; González-Rodríguez, A.; Fernández-Álvarez, A.; Cucarella, C.; Mayoral, R.; Castro-Sánchez, L.; García-Casarrubios, E.; Boscá, L.; et al. Hepatic cyclooxygenase-2 expression protects against diet-induced steatosis, obesity and insulin resistance. Diabetes 2014, 64, 1522–1531. [Google Scholar]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Eichbaum, C.; Meyer, A.S.; Wang, N.; Bischofs, E.; Steinborn, A.; Bruckner, T.; Brodt, P.; Sohn, C.; Eichbaum, M.H.R. Breast cancer cell-derived cytokines, macrophages and cell adhesion: Implications for metastasis. Anticancer Res. 2011, 31, 3219–3227. [Google Scholar] [PubMed]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jae, H.C.; Jong, B.K.; Seok, J.N.; Yang, J.H.; Kim, J.H.; Jeong, E.L. Berberine suppresses TNF-α-induced MMP-9 and cell invasion through inhibition of AP-1 activity in MDA-MB-231 human breast cancer cells. Molecules 2008, 13, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.E.; Schwertfeger, K.L. Proinflammatory cytokines in breast cancer: Mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 2010, 11, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.C.; Panis, C.; Victorino, V.J.; Campos, F.C.; Coloado-Simao, A.N.; Cecchini, A.L.; Cecchini, R. Molecular subtype is determinant on inflammatory status and immunological profile from invasive breast cancer patients. Cancer Immunol. Immunother. 2012, 61, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Turkoz, F.P.; Solak, M.; Petekkaya, I.; Keskin, O.; Kertmen, N.; Sarici, F.; Arik, Z.; Babacan, T.; Ozisik, Y.; Altundag, K. The prognostic impact of obesity on molecular subtypes of breast cancer in premenopausal women. J. BUON 2013, 18, 335–341. [Google Scholar] [PubMed]
- Sethi, S.; Sarkar, F.H.; Ahmed, Q.; Bandyopadhyay, S.; Nahleh, Z.A.; Semaan, A.; Sakr, W.; Munkarah, A.; Ali-Fehmi, R. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl. Oncol. 2011, 4, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Karn, T.; Liedtke, C.; Pusztai, L.; Ruckhaeberle, E.; Hanker, L.; Gaetje, R.; Solbach, C.; Ahr, A.; Metzler, D.; et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011, 13, R97. [Google Scholar] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, E.F.; Sobero, M.E.; Hanauer, D.A.; Sabel, M.S.; Herrmann, E.J.; Weiser, L.J.; Jagielski, C.H.; Griggs, J.J. Obesity and angiolymphatic invasion in primary breast cancer. Ann. Surg. Oncol. 2010, 17, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.; Kundranda, M.N.; Spiro, T.P.; Daw, H.A. The association of metabolic syndrome with triple-negative breast cancer. Breast Cancer Res. Treat. 2010, 121, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Corvera, S.; Gealekman, O. Adipose tissue angiogenesis: Impact on obesity and type-2 diabetes. Biochem. Biophys. Acta 2014, 1842, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, B.; Nicolas, P.; Cucherat, M.; Perret, G.Y. Microvessel density as a prognostic factor in women with breast cancer: A systematic review of the literature and meta-analysis. Cancer Res. 2004, 64, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rose, D.P.; Gracheck, P.J.; Vona-Davis, L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers 2015, 7, 2147-2168. https://doi.org/10.3390/cancers7040883
Rose DP, Gracheck PJ, Vona-Davis L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers. 2015; 7(4):2147-2168. https://doi.org/10.3390/cancers7040883
Chicago/Turabian StyleRose, David P., Peter J. Gracheck, and Linda Vona-Davis. 2015. "The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer" Cancers 7, no. 4: 2147-2168. https://doi.org/10.3390/cancers7040883
APA StyleRose, D. P., Gracheck, P. J., & Vona-Davis, L. (2015). The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers, 7(4), 2147-2168. https://doi.org/10.3390/cancers7040883