
TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Human Fibroblast Isolation and Culturing
2.2. Protein Extraction, Western Blot and Cytokine Array
2.3. Immunofluorescence
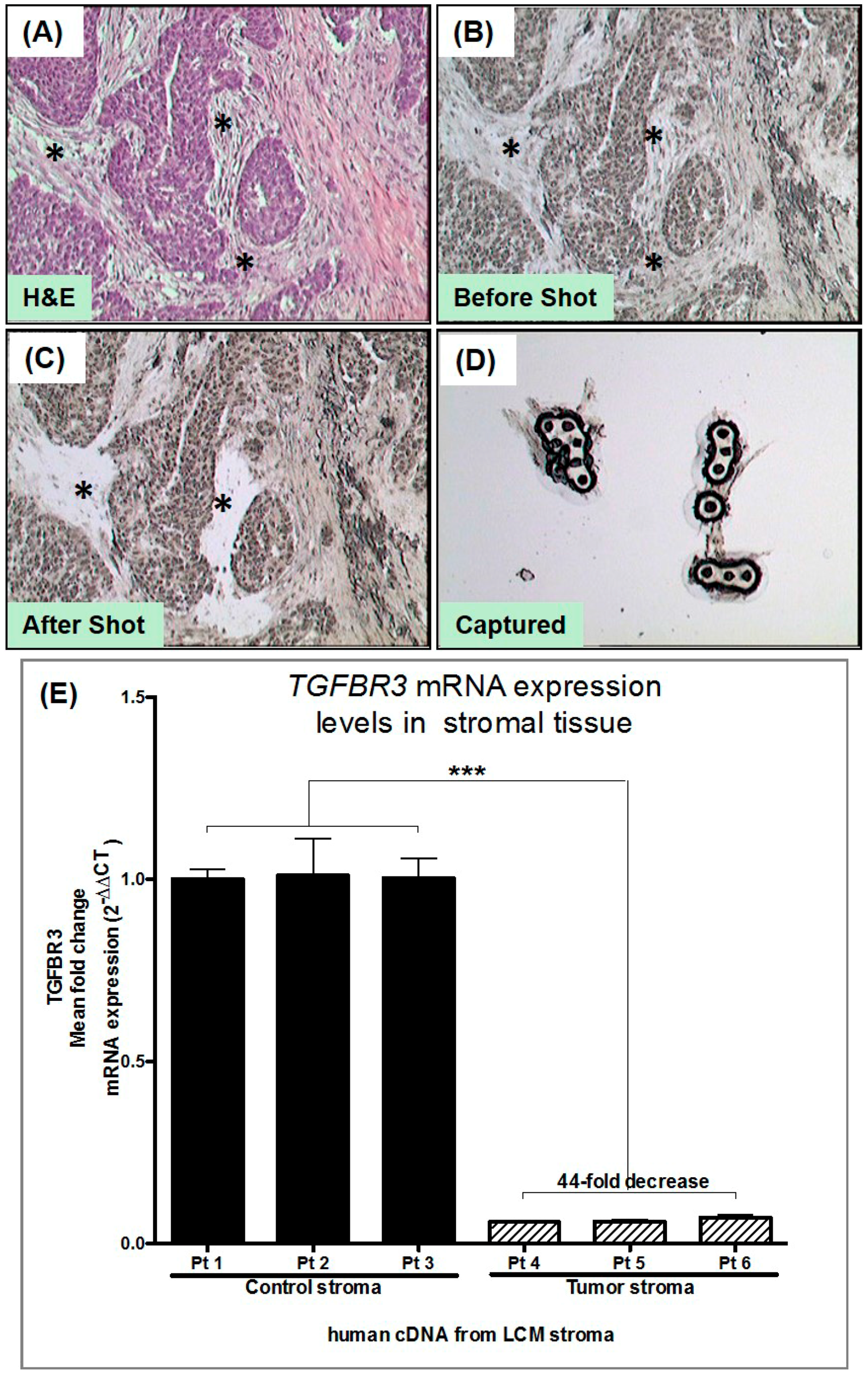
2.4. Laser Capture Microdissection and Expression Analysis
2.5. RNA Preparation and Quantitative PCR (qPCR)
2.6. Statistical Analysis, Bioinformatics and Database Analysis
3. Results
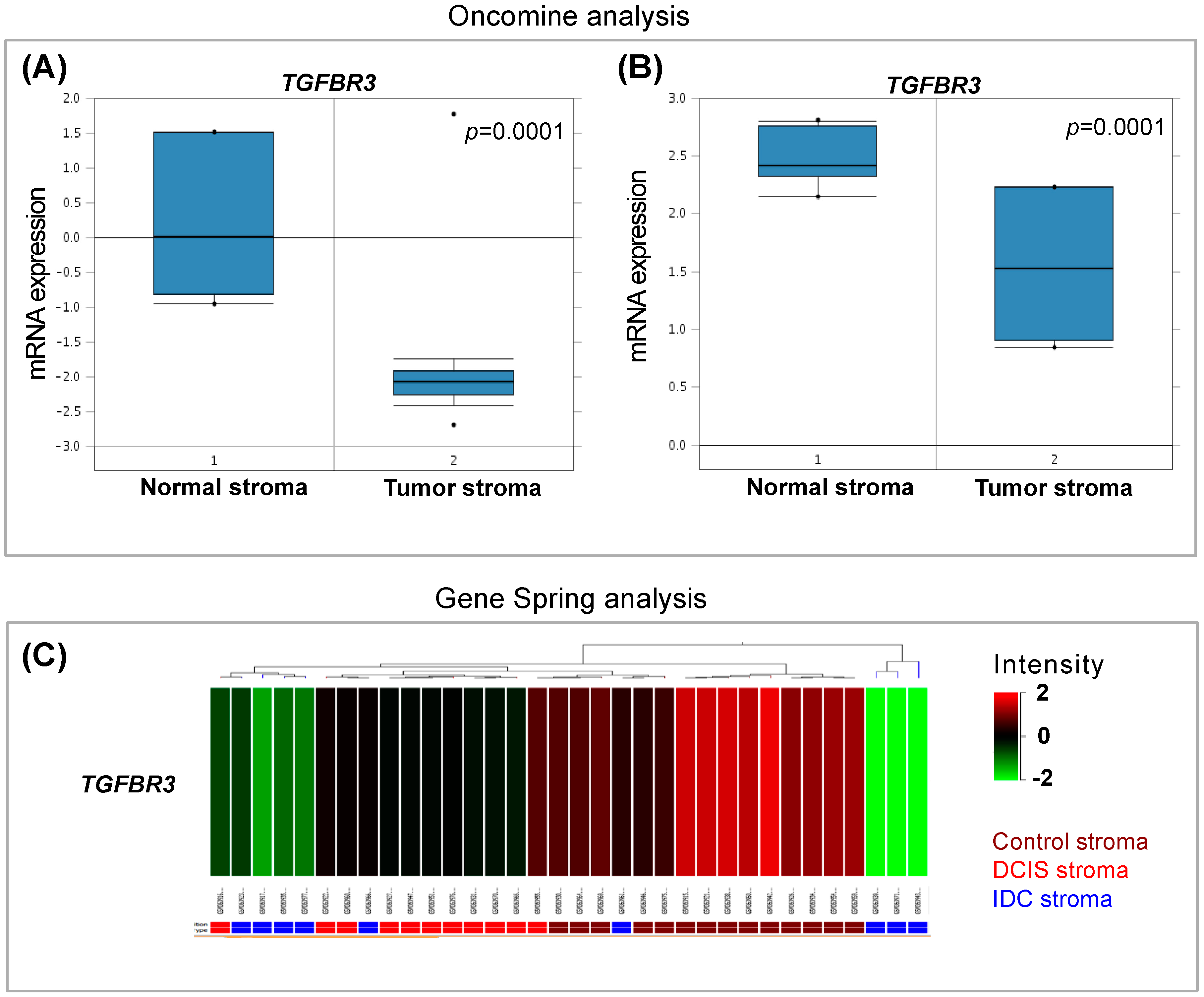
3.1. In Silico Analysis of TGF-β Pathway Related Genes Reveals TGFBR3 to Be Significantly Changed in Tumor Stroma
3.2. Laser Capture Microdissection (LCM) of Human Tumor Stromal Cells
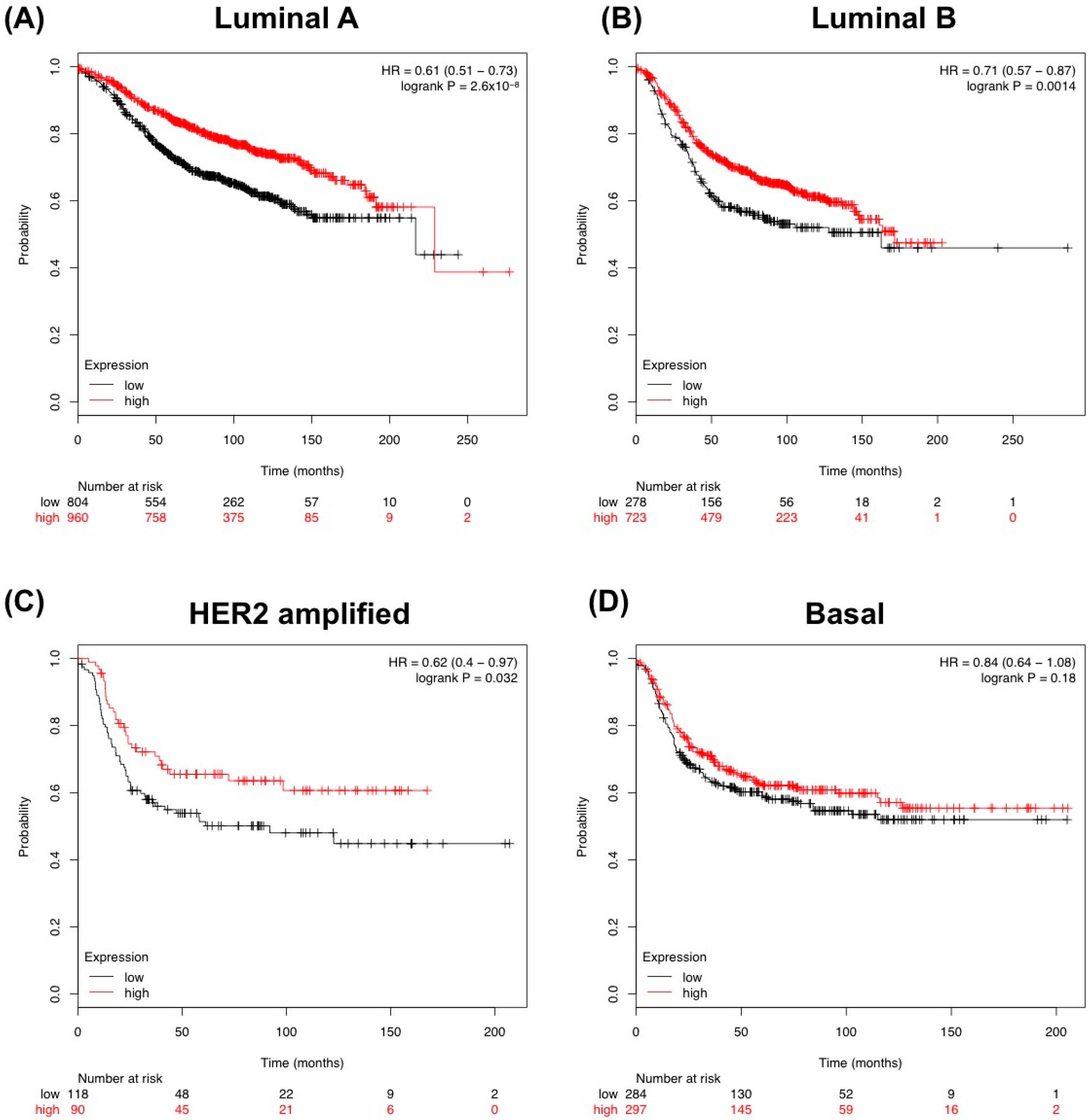
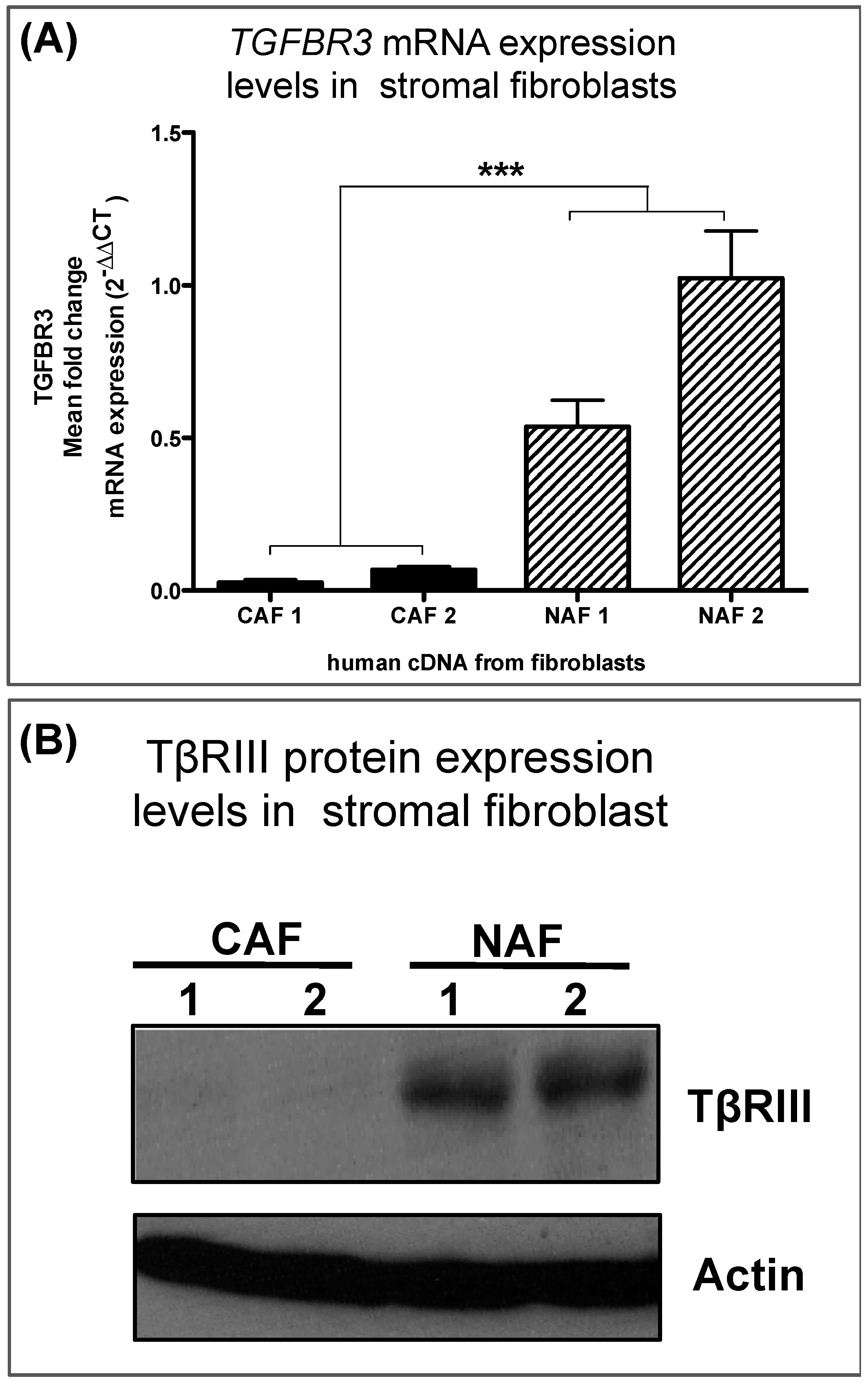
3.3. Human Breast Cancer-Associated Fibroblasts Have Reduced TGFBR3 mRNA and Protein
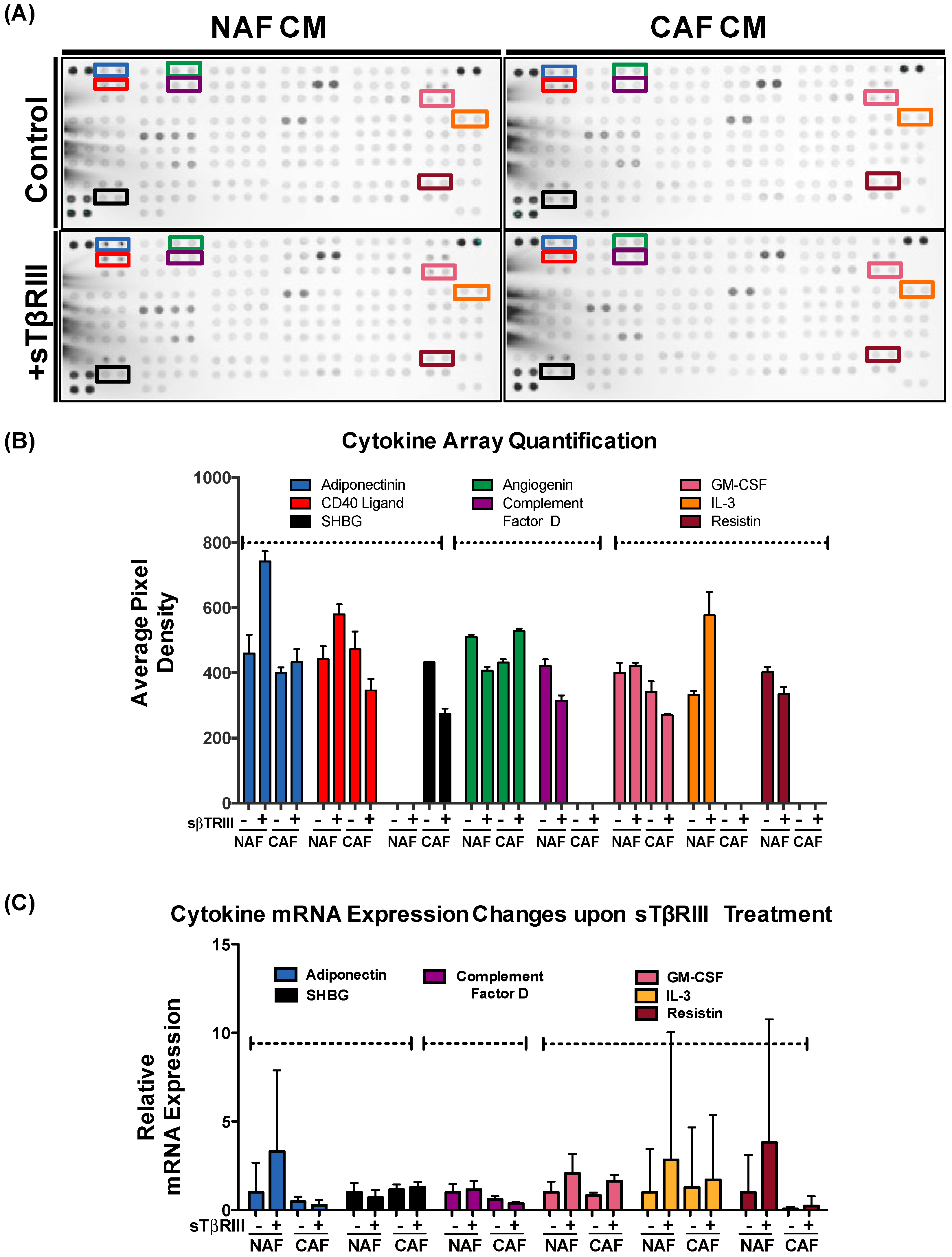
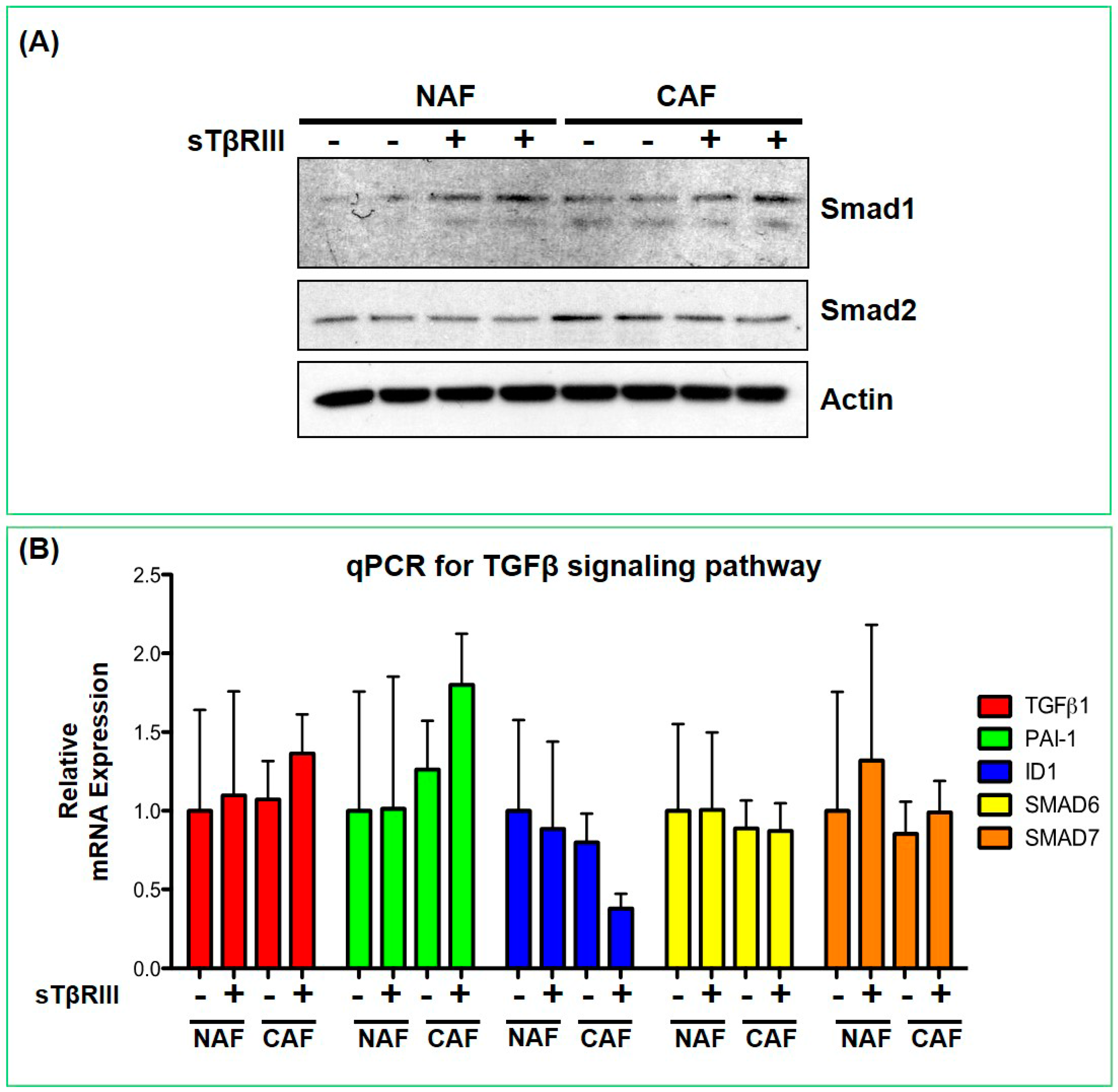
3.4. Human Breast Cancer Associated Fibroblasts and Normal Associated Fibroblasts Have Distinct Inflammatory Responses to Soluble TGFBR3
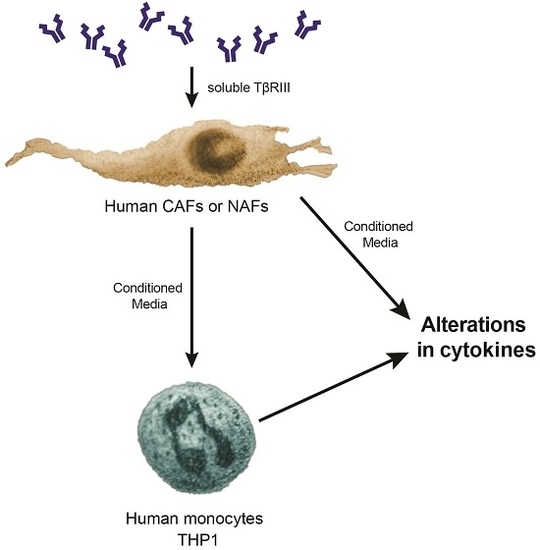
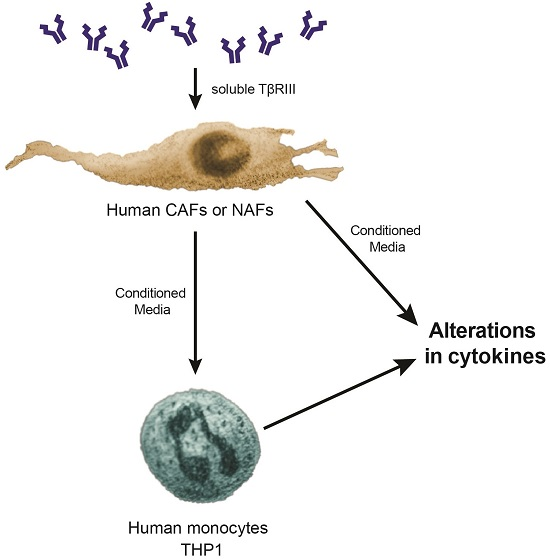
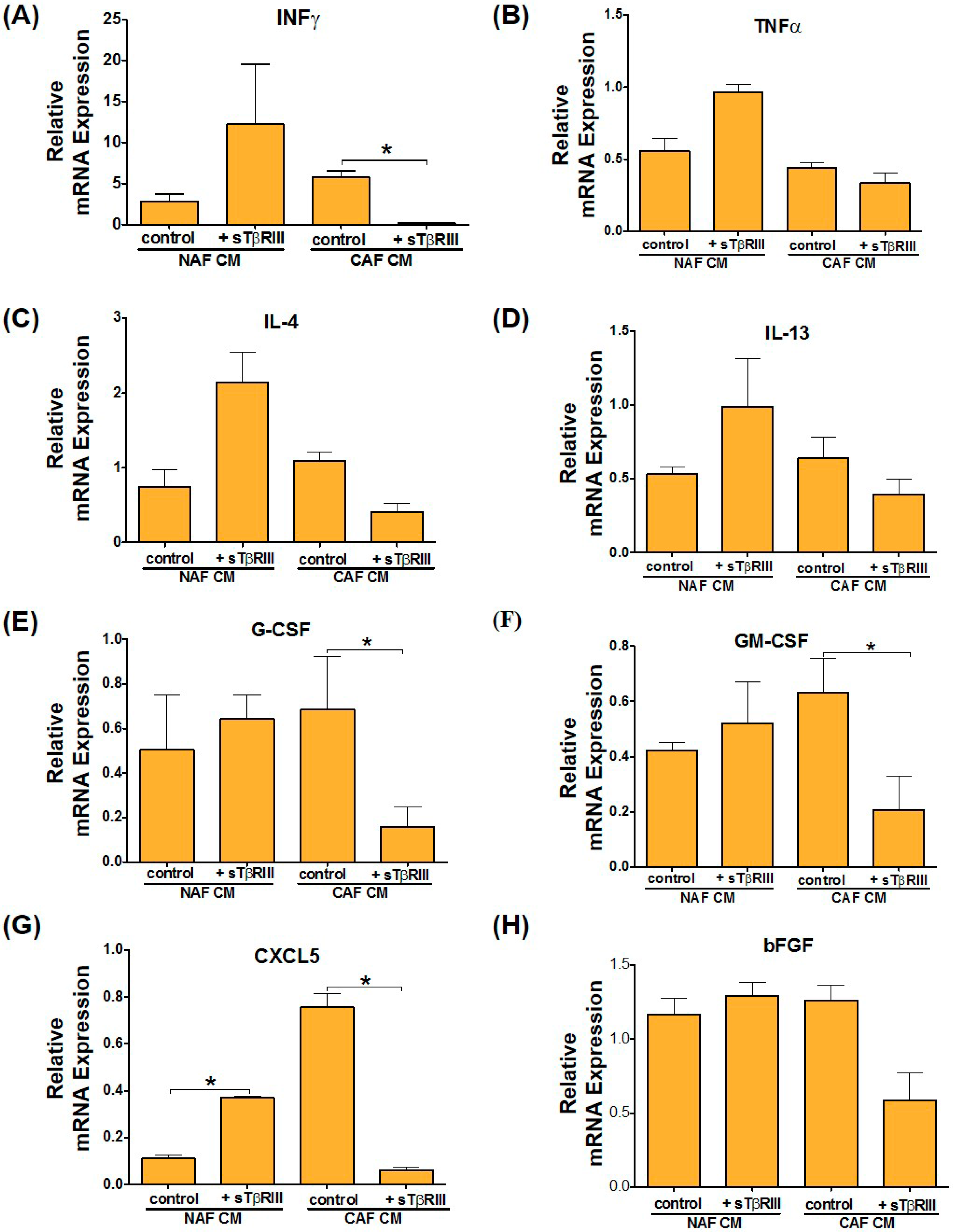
3.5. Treatment of NAFs and CAFs with sTβRIII and Subsequent Conditioned Medium Alters Human Monocytes Cytokine Expression
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- De Kruijf, E.M.; van Nes, J.G.; van de Velde, C.J.; Putter, H.; Smit, V.T.; Liefers, G.J.; Kuppen, P.J.; Tollenaar, R.A.; Mesker, W.E. Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast Cancer Res. Treat. 2011, 125, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Bhowmick, N.A.; Chytil, A.; Gorksa, A.E.; Brown, K.A.; Muraoka, R.; Arteaga, C.L.; Neilson, E.G.; Hayward, S.W.; Moses, H.L. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP- and HGF-mediated signaling networks. Oncogene 2005, 24, 5053–5068. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Enhanced hepatocyte growth factor signaling by type II transforming growth factor-β receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res. 2007, 67, 4869–4877. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Transforming growth factor-β signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res. 2008, 6, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Laklai, H.; Acerbi, I.; Owens, P.; Gorska, A.E.; Chytil, A.; Aakre, M.; Weaver, V.M.; Moses, H.L. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas. Cancer Res. 2013, 73, 5336–5346. [Google Scholar] [CrossRef] [PubMed]
- Owens, P.; Polikowsky, H.; Pickup, M.W.; Gorska, A.E.; Jovanovic, B.; Shaw, A.K.; Novitskiy, S.V.; Hong, C.C.; Moses, H.L. Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PLoS ONE 2013, 8, e67533. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Hover, L.D.; Polikowsky, E.R.; Chytil, A.; Gorska, A.E.; Novitskiy, S.V.; Moses, H.L.; Owens, P. Bmpr2 loss in fibroblasts promotes mammary carcinoma metastasis via increased inflammation. Mol. Oncol. 2015, 9, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.K.; Pickup, M.W.; Chytil, A.; Aakre, M.; Owens, P.; Moses, H.L.; Novitskiy, S.V. Tgfbeta signaling in myeloid cells regulates mammary carcinoma cell invasion through fibroblast interactions. PLoS ONE 2015, 10, e0117908. [Google Scholar] [CrossRef] [PubMed]
- Hanks, B.A.; Holtzhausen, A.; Evans, K.S.; Jamieson, R.; Gimpel, P.; Campbell, O.M.; Hector-Greene, M.; Sun, L.; Tewari, A.; George, A.; et al. Type III TGF-β receptor downregulation generates an immunotolerant tumor microenvironment. J. Clin. Investig. 2013, 123, 3925–3940. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; How, T.; Kirkbride, K.C.; Gordon, K.J.; Lee, J.D.; Hempel, N.; Kelly, P.; Moeller, B.J.; Marks, J.R.; Blobe, G.C. The type III TGF-β receptor suppresses breast cancer progression. J. Clin. Investig. 2007, 117, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Criswell, T.; Dumont, N.; Barnett, J.; Arteaga, C. Knockdown of the transforming growth factor-β type III receptor impairs motility and invasion of metastatic cancer cells. Cancer Res. 2008, 68, 7304–7312. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuulasmaa, T.; Kosma, V.M.; Butzow, R.; Vanttinen, T.; Hyden-Granskog, C.; Voutilainen, R. Expression of betaglycan, an inhibin coreceptor, in normal human ovaries and ovarian sex cord-stromal tumors and its regulation in cultured human granulosa-luteal cells. J. Clin. Endocrinol. Metab. 2003, 88, 5002–5008. [Google Scholar] [CrossRef] [PubMed]
- Woszczyk, D.; Gola, J.; Jurzak, M.; Mazurek, U.; Mykala-Ciesla, J.; Wilczok, T. Expression of TGF β 1 genes and their receptor types I, II, and III in low- and high-grade malignancy non-hodgkin’s lymphomas. Med. Sci. Monit. 2004, 10, CR33–CR37. [Google Scholar] [PubMed]
- Jelinek, D.F.; Tschumper, R.C.; Stolovitzky, G.A.; Iturria, S.J.; Tu, Y.; Lepre, J.; Shah, N.; Kay, N.E. Identification of a global gene expression signature of B-chronic lymphocytic leukemia. Mol. Cancer Res. 2003, 1, 346–361. [Google Scholar] [PubMed]
- Jovanovic, B.; Beeler, J.S.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Ashby, W.J.; Lehmann, B.D.; Zijlstra, A.; Pietenpol, J.A.; Moses, H.L. Transforming growth factor beta receptor type III is a tumor promoter in mesenchymal-stem like triple negative breast cancer. Breast Cancer Res. 2014, 16, R69. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in caf-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pardali, K.; Moustakas, A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 2007, 1775, 21–62. [Google Scholar] [PubMed]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGF beta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGF beta: The molecular jekyll and hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009, 11, R7. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rasanen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Acar, A.; Magnusson, Y.; Gregersson, P.; Ryden, L.; Landberg, G. Tgf-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene 2015, 34, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Criswell, T.L.; Arteaga, C.L. Modulation of nfkappab activity and e-cadherin by the type III transforming growth factor beta receptor regulates cell growth and motility. J. Biol. Chem. 2007, 282, 32491–32500. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Dong, M.; Chislock, E.M.; Fields, T.A.; Blobe, G.C. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis 2008, 29, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Loyden, G.; Arribas, J.; Lopez-Casillas, F. The shedding of betaglycan is regulated by pervanadate and mediated by membrane type matrix metalloprotease-1. J. Biol. Chem. 2004, 279, 7721–7733. [Google Scholar] [CrossRef] [PubMed]
- Turley, R.S.; Finger, E.C.; Hempel, N.; How, T.; Fields, T.A.; Blobe, G.C. The type III transforming growth factor-β receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007, 67, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Finger, E.C.; Lee, N.Y.; You, H.J.; Blobe, G.C. Endocytosis of the type III transforming growth factor-β (TGF-beta) receptor through the clathrin-independent/lipid raft pathway regulates TGF-beta signaling and receptor down-regulation. J. Biol. Chem. 2008, 283, 34808–34818. [Google Scholar] [CrossRef] [PubMed]
- Mythreye, K.; Blobe, G.C. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc. Natl. Acad. Sci. USA 2009, 106, 8221–8226. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.E.; Huang, H.; Mythreye, K.; Blobe, G.C. The type III transforming growth factor-β receptor inhibits proliferation, migration, and adhesion in human myeloma cells. Mol. Biol. Cell 2011, 22, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of tgf-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 2009, 1792, 954–973. [Google Scholar] [CrossRef] [PubMed]
- Dotor, J.; Lopez-Vazquez, A.B.; Lasarte, J.J.; Sarobe, P.; Garcia-Granero, M.; Riezu-Boj, J.I.; Martinez, A.; Feijoo, E.; Lopez-Sagaseta, J.; Hermida, J.; et al. Identification of peptide inhibitors of transforming growth factor beta 1 using a phage-displayed peptide library. Cytokine 2007, 39, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Wang, H.; Zhou, J.; Li, B.; Zhang, J.; Lu, M.; Wang, Z. P144, a TGF-beta1 antagonist peptide, synergizes with sildenafil and enhances erectile response via amelioration of cavernosal fibrosis in diabetic rats. J. Sex. Med. 2013, 10, 2942–2951. [Google Scholar] [CrossRef] [PubMed]
- San-Martin, A.; Dotor, J.; Martinez, F.; Hontanilla, B. Effect of the inhibitor peptide of the transforming growth factor beta (P144) in a new silicone pericapsular fibrotic model in pigs. Aesthetic Plast. Surg. 2010, 34, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Zubeldia, I.G.; Bleau, A.M.; Redrado, M.; Serrano, D.; Agliano, A.; Gil-Puig, C.; Vidal-Vanaclocha, F.; Lecanda, J.; Calvo, A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1-targeting peptides P17 and P144. Exp. Cell Res. 2013, 319, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Zarranz-Ventura, J.; Fernandez-Robredo, P.; Recalde, S.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-β inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS ONE 2013, 8, e65434. [Google Scholar] [CrossRef] [PubMed]
- Recalde, S.; Zarranz-Ventura, J.; Fernandez-Robredo, P.; Garcia-Gomez, P.J.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-β inhibition decreases diode laser-induced choroidal neovascularization development in rats: P17 and P144 peptides. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7090–7097. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | FWD SEQUENCE | REV SEQUENCE |
|---|---|---|
| TGFBR3 | TGGGGTCTCCAGACTGTTTTT | CTGCTCCATACTCTTTTCCGGG |
| ADIPONECTIN | AACATGCCCATTCGCTTTACC | TAGGCAAAGTAGTACAGCCCA |
| COMPLEMENT FACTOR D | GACACCATCGACCACGACC | GCCACGTCGCAGAGAGTTC |
| GM-CSF | TCCTGAACCTGAGTAGAGACAC | TGCTGCTTGTAGTGGCTGG |
| IL-3 | TCAACAGGGCTGTCAAGAGTT | CAGATAGAACGTCAGTTTCCTCC |
| RESISTIN | CTGTTGGTGTCTAGCAAGACC | CCAATGCTGCTTATTGCCCTAAA |
| SHBG | GCCCAGGACAAGAGCCTATC | CCTTAGGGTTGGTATCCCCATAA |
| INFγ | TCGGTAACTGACTTGAATGTCCA | TCGCTTCCCTGTTTTAGCTGC |
| TNFα | GAGGCCAAGCCCTGGTATG | CGGGCCGATTGATCTCAGC |
| IL-4 | CGGCAACTTTGTCCACGGA | TCTGTTACGGTCAACTCGGTG |
| IL-13 | GAAGGCTCCGCTCTGCAAT | TCCAGGGCTGCACAGTACA |
| G-CSF | GCTGCTTGAGCCAACTCCATA | GAACGCGGTACGACACCTC |
| CXCL5 | AGCTGCGTTGCGTTTGTTTAC | TGGCGAACACTTGCAGATTAC |
| bFGF | AGAAGAGCGACCCTCACATCA | CGGTTAGCACACACTCCTTTG |
| TGFβ1 | CAATTCCTGGCGATACCTCAG | GCACAACTCCGGTGACATCAA |
| PAI-1 | GACATCCTGGAACTGCCCTA | GGTCATGTTGCCTTTCCAAGT |
| ID1 | CTGCTCTACGACATGAACGG | GAAGGTCCCTGATGTAGTCGAT |
| SMAD6 | CCTCCCTACTCTCGGCTGTC | GGTAGCCTCCGTTTCAGTGTA |
| SMAD7 | CCAACTGCAGACTGTCCAGA | CAGGCTCCAGAAGAAGTTGG |
| GAPDH | CTGGGCTACACTGAGCACC | AAGTGGTCGTTGAGGGCAATG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanović, B.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Johnson, K.C.; Moses, H.L.; Owens, P. TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts. Cancers 2016, 8, 100. https://doi.org/10.3390/cancers8110100
Jovanović B, Pickup MW, Chytil A, Gorska AE, Johnson KC, Moses HL, Owens P. TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts. Cancers. 2016; 8(11):100. https://doi.org/10.3390/cancers8110100
Chicago/Turabian StyleJovanović, Bojana, Michael W. Pickup, Anna Chytil, Agnieszka E. Gorska, Kimberly C. Johnson, Harold L. Moses, and Philip Owens. 2016. "TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts" Cancers 8, no. 11: 100. https://doi.org/10.3390/cancers8110100
APA StyleJovanović, B., Pickup, M. W., Chytil, A., Gorska, A. E., Johnson, K. C., Moses, H. L., & Owens, P. (2016). TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts. Cancers, 8(11), 100. https://doi.org/10.3390/cancers8110100