
Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment


Abstract
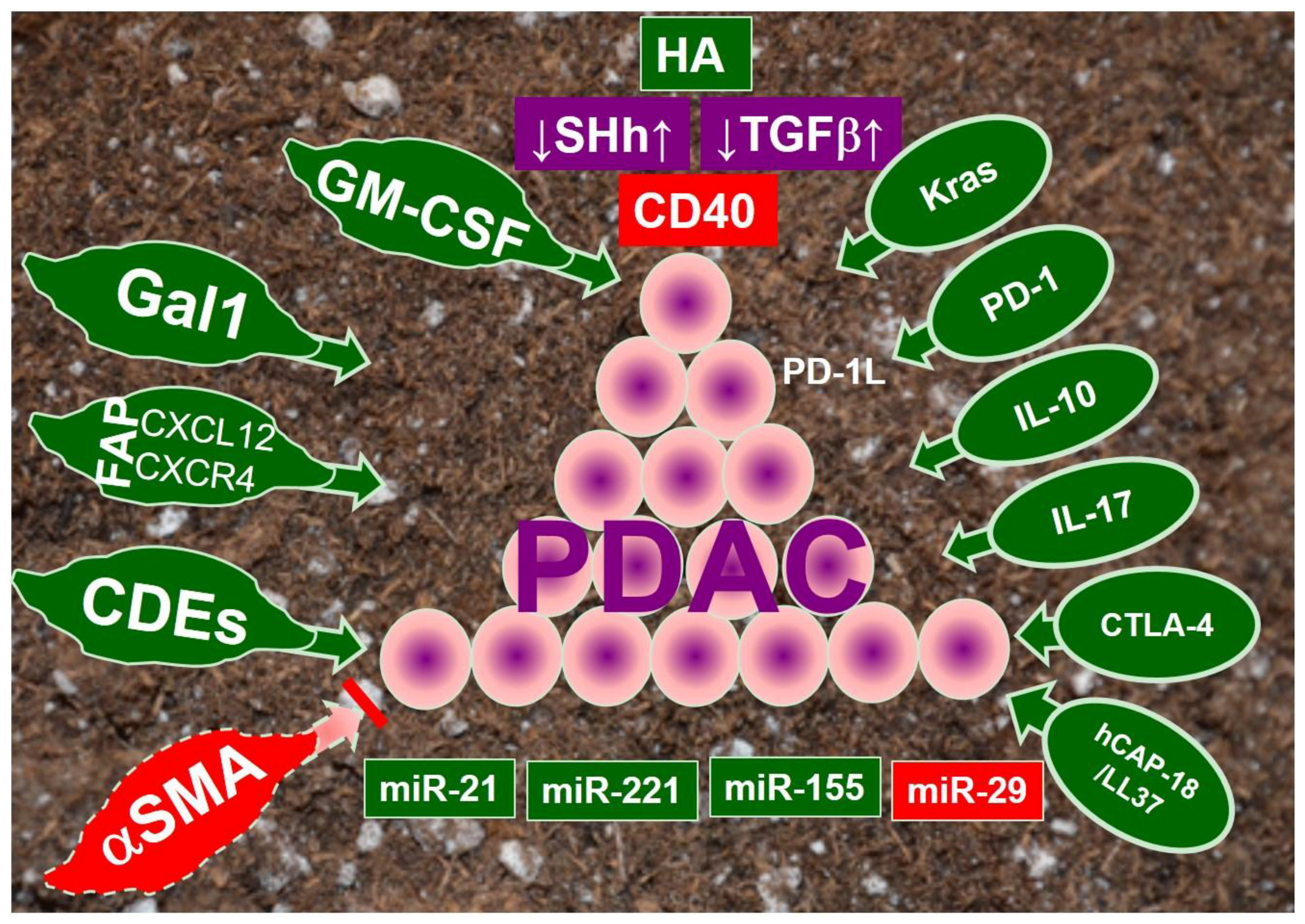
:1. Introduction
2. Cancer-Associated Fibroblasts
3. Immune Modulation
4. Signaling Pathways in PDAC Stroma
4.1. Hyaluronan
4.2. Sonic Hedgehog
4.3. Transforming Growth Factor β (TGFβ)
4.4. Abberrant Immune Regulators in Tumor Microenvironment
4.5. Constitutively Activated Kras Pathway
5. MicroRNAs
6. Cancer Vaccines
7. Concluding Remarks and Future Treatment Regimens
Acknowledgments
Conflicts of Interest
References
- Schneider, G.; Schmid, R.M. Genetic alterations in pancreatic carcinoma. Mol. Cancer 2003, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed]
- Moir, J.A.; White, S.A.; Mann, J. Arrested development and the great escape—the role of cellular senescence in pancreatic cancer. Int. J. Biochem. Cell Biol. 2014, 57, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Albo, D.; Berger, D.H. The role of the tumor microenvironment in the progression of pancreatic cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Zilberstein, Y.; Lavy, A.; Nagler, A.; Genin, O.; Pines, M. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS ONE 2012, 7, e41833. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancrea 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Fernandez-Barrena, M.G.; Moreno, M.; Ortiz-Zapater, E.; Munne-Collado, J.; Iglesias, M.; Andre, S.; Gabius, H.J.; Hwang, R.F.; Poirier, F.; et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res. 2014, 74, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.C.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Bramhall, S.R.; Rosemurgy, A.; Brown, P.D.; Bowry, C.; Buckels, J.A.; Marimastat Pancreatic Cancer Study Group. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: A randomized trial. J. Clin. Oncol. 2001, 19, 3447–3455. [Google Scholar] [CrossRef] [PubMed]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Hamm, J.; Dancey, J.; Eisenberg, P.D.; Dagenais, M.; Fields, A.; Hagan, K.; Greenberg, B.; Colwell, B.; Zee, B.; et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Gore, J.; Korc, M. Pancreatic cancer stroma: Friend or foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.P.; De Monte, L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol. 2013, 4, 210. [Google Scholar] [CrossRef] [PubMed]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Schultze, J.L. Regulatory T cells in cancer. Blood 2006, 108, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Howie, D.; Nolan, K.F.; Agorogiannis, E.I.; Greaves, D.R.; Cobbold, S.P.; Waldmann, H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-beta. Eur. J. Immunol. 2011, 41, 2955–2965. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Hwang-Verslues, W.W.; Lee, W.H.; Huang, C.K.; Wei, P.C.; Chen, C.L.; Shew, J.Y.; Lee, E.Y.; Jeng, Y.M.; Tien, Y.W.; et al. Targeting IL-17B-IL-17RB signaling with an anti-IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J. Exp. Med. 2015, 212, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; Lopez-Millan, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 Functions through the Novel REG3beta-JAK2-STAT3 Inflammatory Pathway to Promote the Transition from Chronic Pancreatitis to Pancreatic Cancer. Cancer Res. 2015, 75, 4852–4862. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, C.; Li, W.; Gao, Z.; Guo, K.; Song, S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life 2014, 66, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Leone, B.E.; Zerbi, A.; Balzano, G.; Cainarca, S.; Sordi, V.; Pontillo, M.; Mercalli, A.; Di Carlo, V.; Allavena, P.; et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J. Immunol. 2004, 172, 7341–7349. [Google Scholar] [CrossRef] [PubMed]
- Karnevi, E.; Andersson, R.; Rosendahl, A.H. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 2014, 92, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Kruger, U.; Becker, T.; Ebsen, M.; Rocken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer. 2014, 135, 843–861. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chheda, C.; Ouhaddi, Y.; Benhaddou, H.; Bourhim, M.; Grippo, P.J.; Principe, D.R.; Mascarinas, E.; DeCant, B.; Tsukamoto, H.; et al. Characterization of Mouse Models of Early Pancreatic Lesions Induced by Alcohol and Chronic Pancreatitis. Pancreas 2015, 44, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Alcala, S.; Garcia, E.; Sanchez-Ripoll, Y.; Azevedo, M.M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; Gomez-Lopez, G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Gironella, M.; Calvo, C.; Fernandez, A.; Closa, D.; Iovanna, J.L.; Rosello-Catafau, J.; Folch-Puy, E. Reg3beta deficiency impairs pancreatic tumor growth by skewing macrophage polarization. Cancer Res. 2013, 73, 5682–5694. [Google Scholar] [CrossRef] [PubMed]
- Michl, P.; Gress, T.M. Improving drug delivery to pancreatic cancer: Breaching the stromal fortress by targeting hyaluronic acid. Gut 2012, 61, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, Q.; Duan, W.; Liu, H.; Xu, H.; Wu, E. Paracrine sonic hedgehog signaling derived from tumor epithelial cells: A key regulator in the pancreatic tumor microenvironment. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; Pasca di Magliano, M. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-beta2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Duewell, P. Breaking tumor-induced immunosuppression with 5′-triphosphate siRNA silencing TGFbeta and activating RIG-I. Oncoimmunology 2013, 2, e24170. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Sato, K.; Yamazaki, Y.; Ohyama, T.; Horiguchi, N.; Hashizume, H.; Kakizaki, S.; Takagi, H.; Ozaki, I.; Arai, H.; et al. Transforming growth factor-alpha activates pancreatic stellate cells and may be involved in matrix metalloproteinase-1 upregulation. Lab. Investig. 2013, 93, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Ostapoff, K.T.; Cenik, B.K.; Wang, M.; Ye, R.; Xu, X.; Nugent, D.; Hagopian, M.M.; Topalovski, M.; Rivera, L.B.; Carroll, K.D.; et al. Neutralizing murine TGFbetaR2 promotes a differentiated tumor cell phenotype and inhibits pancreatic cancer metastasis. Cancer Res. 2014, 74, 4996–5007. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sun, X.; Qian, K.Q.; Liu, X.; Wang, Z.; Chen, Y. Protection of cerulein-induced pancreatic fibrosis by pancreas-specific expression of Smad7. Biochim. Biophys. Acta 2009, 1792, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Gibbons, D.L.; Kurie, J.M. The role of epithelial-mesenchymal transition programming in invasion and metastasis: A clinical perspective. Cancer Manag. Res. 2013, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.A.; Merchant, N.B.; Nagathihalli, N.S. Emerging targets in pancreatic cancer: Epithelial-mesenchymal transition and cancer stem cells. Onco Targets Ther. 2013, 6, 1261–1267. [Google Scholar] [PubMed]
- Sotomayor, E.M.; Borrello, I.; Tubb, E.; Rattis, F.M.; Bien, H.; Lu, Z.; Fein, S.; Schoenberger, S.; Levitsky, H.I. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 1999, 5, 780–787. [Google Scholar] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Lohr, M.; Kloppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Luttges, J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. Role of RAS in the regulation of PI 3-kinase. Curr. Top. Microbiol. Immunol. 2010, 346, 143–169. [Google Scholar] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; Pasca di Magliano, M.; Fernandez-Zapico, M.E. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 2013, 288, 11786–11794. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.P.; Reginato, M.J.; Reichert, M.; Rustgi, A.K.; Lelkes, P.I. Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1. Mol. Cancer Res. 2012, 10, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Suresh, R.; Banerjee, S.; Bao, B.; Xu, Z.; Wilson, J.; Philip, P.A.; Apte, M.; Sarkar, F.H. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am. J. Cancer Res. 2015, 5, 1251–1264. [Google Scholar] [PubMed]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.J.; Nabinger, S.C.; Vega, Z.; Sahu, S.S.; Alluri, R.K.; Abdul-Sater, Z.; Yu, Z.; Gore, J.; Nalepa, G.; Saxena, R.; et al. Pathophysiological role of microRNA-29 in pancreatic cancer stroma. Sci. Rep. 2015, 5, 11450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.F.; Zhou, Q.B.; Liao, Y.D.; Mai, C.; Chen, T.J.; Tang, Y.Q.; Chen, R.F. Optimized construction of MUC1-VNTRn DNA vaccine and its anti-pancreatic cancer efficacy. Oncol. Lett. 2017, 13, 2198–2206. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Target | Clinical Studies | Reference |
|---|---|---|
| Hyaluronan and chemotherapy agent | Phase 1b PEGPH20 plus Gemcitabine | [50] |
| Transforming Growth Factor β | Orthotopic human tumor xenografts TGFβr2 neutralizing antibody (2G8) | [62] |
| Cancer vaccine | GVAX. Phase 2 | [83,84] |
| Cancer vaccine and CTLA-4 | GVAX and ipilimumab | [85] |
| Cancer vaccine and Treg | GVAX and cyclophosphamide | [86] |
| Cancer vaccine, Treg, and mesothelin | Cyclophosphamide/GVAXCRS-207 | [87] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-J.; Lin, J. Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment. Cancers 2017, 9, 93. https://doi.org/10.3390/cancers9070093
Lin H-J, Lin J. Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment. Cancers. 2017; 9(7):93. https://doi.org/10.3390/cancers9070093
Chicago/Turabian StyleLin, Huey-Jen, and Jiayuh Lin. 2017. "Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment" Cancers 9, no. 7: 93. https://doi.org/10.3390/cancers9070093
APA StyleLin, H. -J., & Lin, J. (2017). Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment. Cancers, 9(7), 93. https://doi.org/10.3390/cancers9070093