
Radioresistance of Brain Tumors

{kind=link}
Abstract
:1. Introduction
1.1. Radiotherapy and Radioresistance of Brain Tumors
1.2. Effects of Radiation on Normal Brain Tissue
2. Dose Escalation and Altered Fractionation to Combat Radioresistance
3. Rationale for Pharmaceutical Radiosensitizers
4. Tumor Heterogeneity
5. Enrichment of CSC by Ionizing Radiation (IR)
6. DNA Damage Response
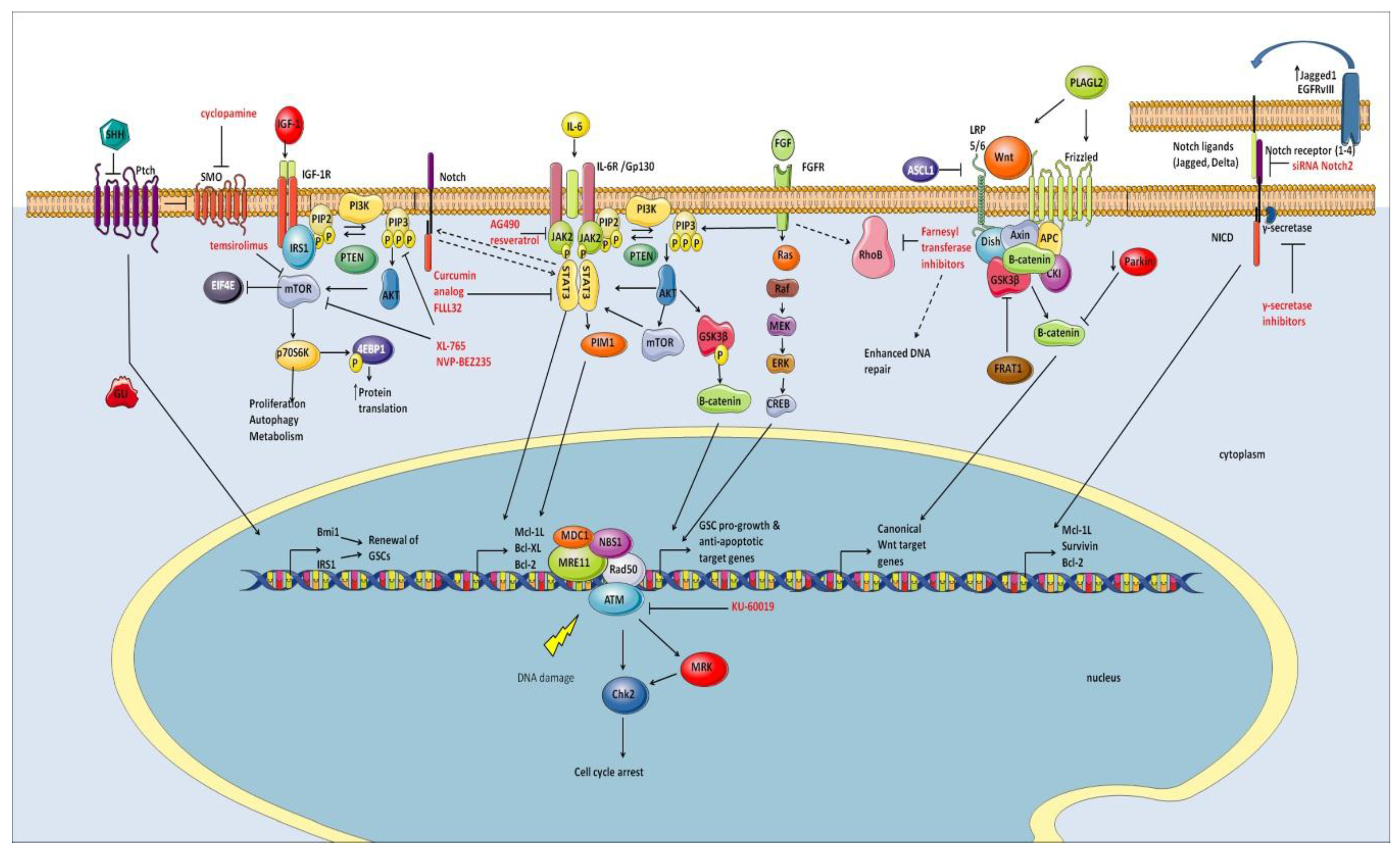
7. Signaling Pathways
7.1. Notch
7.2. Wnt/β-Catenin Pathway
7.3. SHH/Gli Pathway
7.4. FGF-2
7.5. EGFR
7.6. IGF
7.7. cMet
7.8. PI3K/Akt Pathway
7.9. mTOR
7.10. NF-κB
7.11. STAT3
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brandsma, D.; Stalpers, L.; Taal, W.; Sminia, P.; van Den Bent, M.J. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 2008, 9, 453–461. [Google Scholar] [CrossRef]
- Siu, A.; Wind, J.J.; Iorgulescu, J.B.; Chan, T.A.; Yamada, Y.; Sherman, J.H. Radiation necrosis following treatment of high grade glioma—A review of the literature and current understanding. Acta Neurochir. (Wien). 2012, 154, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Chen, P.; Haimovitz-Friedman, A.; Reilly, R.M.; Wong, C.S. Endothelial apoptosis initiates acute blood-brain barrier disruption after ionizing radiation. Cancer Res. 2003, 63, 5950–5956. [Google Scholar] [PubMed]
- Chiang, C.S.; Hong, J.H.; Stalder, A.; Sun, J.R.; Withers, H.R.; McBride, W.H. Delayed molecular responses to brain irradiation. Int. J. Radiat. Biol. 1997, 72, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Chiang, C.S.; Campbell, I.L.; Sun, J.R.; Withers, H.R.; McBride, W.H. Induction of acute phase gene expression by brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 619–626. [Google Scholar] [CrossRef]
- Rodel, F.; Frey, B.; Multhoff, G.; Gaipl, U. Contribution of the immune system to bystander and non-targeted effects of ionizing radiation. Cancer Lett. 2015, 356, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Sprung, C.N.; Ivashkevich, A.; Forrester, H.B.; Redon, C.E.; Georgakilas, A.; Martin, O.A. Oxidative DNA damage caused by inflammation may link to stress-induced non-targeted effects. Cancer Lett. 2015, 356, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Havaki, S.; Kotsinas, A.; Chronopoulos, E.; Kletsas, D.; Georgakilas, A.; Gorgoulis, V.G. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Lett. 2015, 356, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Martin, M.; Linan, O.; Alvarenga, F.; Lopez, M.; Fernandez, L.; Buchser, D.; Cerezo, L. Bystander effects and radiotherapy. Rep. Pract. Oncol. Radiother. 2015, 20, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zelefsky, M.J.; Pei, X.; Chou, J.F.; Schechter, M.; Kollmeier, M.; Cox, B.; Yamada, Y.; Fidaleo, A.; Sperling, D.; Happersett, L.; et al. Dose escalation for prostate cancer radiotherapy: Predictors of long-term biochemical tumor control and distant metastases-free survival outcomes. Eur. Urol. 2011, 60, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Sato, K.; Horie, A.; Iketani, S.; Yamada, H.; Nakatani, Y.; Sato, J.; Hamada, Y. Stereotactic radiosurgery may contribute to overall survival for patients with recurrent head and neck carcinoma. Radiat. Oncol. 2010, 5, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.R.; Lominska, C.E.; Deeken, J.F.; Davidson, B.J.; Newkirk, K.A.; Gagnon, G.J.; Hwang, J.; Slack, R.S.; Noone, A.M.; Harter, K.W. Fractionated stereotactic radiosurgery for reirradiation of head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Khoo, V.S. Radiotherapeutic techniques for prostate cancer, dose escalation and brachytherapy. Clin. Oncol. 2005, 17, 560–571. [Google Scholar] [CrossRef]
- Kuban, D.A.; Tucker, S.L.; Dong, L.; Starkschall, G.; Huang, E.H.; Cheung, M.R.; Lee, A.K.; Pollack, A. Long-Term results of the MD Anderson randomized dose-escalation trial for prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, R.; Paulus, R.; Galvin, J.; Michalski, J.; Straube, W.; Bradley, J.; Fakiris, A.; Bezjak, A.; Videtic, G.; Johnstone, D.; et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA 2010, 303, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Tsien, C.; Moughan, J.; Michalski, J.M.; Gilbert, M.R.; Purdy, J.; Simpson, J.; Kresel, J.J.; Curran, W.J.; Diaz, A.; Mehta, M.P. Phase I three-dimensional conformal radiation dose escalation study in newly diagnosed glioblastoma: Radiation Therapy Oncology Group trial 98-03. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Randomized Phase II Trial of Hypofractionated Dose-Escalated Photon IMRT or Proton Beam Therapy Versus Conventional Photon Irradiation With Concomitant and Adjuvant Temozolomide in Patients With Newly Diagnosed Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02179086 (accessed on 25 March 2016).
- Prasanna, A.; Ahmed, M.M.; Mohiuddin, M.; Coleman, C.N. Exploiting sensitization windows of opportunity in hyper and hypofractionated radiation therapy. J. Thorac. Dis. 2014, 6, 287–302. [Google Scholar] [PubMed]
- Lee, D.Y.; Chunta, J.L.; Park, S.S.; Huang, J.; Martinez, A.A.; Grills, I.S.; Krueger, S.A.; Wilson, G.D.; Marples, B. Pulsed versus conventional radiation therapy in combination with temozolomide in a murine orthotopic model of glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P.; Bernier, V.; Carnin, C.; Taillandier, L.; Djabri, M.; Martin, L.; Michel, X.; Maire, J.P.; Khalil, T.; Kerr, C.; et al. Prolonged survival for patients with newly diagnosed, inoperable glioblastoma with 3-times daily ultrafractionated radiation therapy. Neuro-oncology 2010, 12, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Leder, K.; Pitter, K.; Laplant, Q.; Hambardzumyan, D.; Ross, B.D.; Chan, T.A.; Holland, E.C.; Michor, F. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell 2014, 156, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Simpson, W.J.; Platts, M.E. Fractionation study in the treatment of glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 1976, 1, 639–644. [Google Scholar] [CrossRef]
- Payne, D.G.; Simpson, W.J.; Keen, C.; Platts, M.E. Malignant astrocytoma: Hyperfractionated and standard radiotherapy with chemotherapy in a randomized prospective clinical trial. Cancer 1982, 50, 2301–2306. [Google Scholar] [CrossRef]
- Coughlin, C.; Scott, C.; Langer, C.; Coia, L.; Curran, W.; Rubin, P. Phase II, two-arm RTOG trial (94-11) of bischloroethyl-nitrosourea plus accelerated hyperfractionated radiotherapy (64.0 or 70.4 Gy) based on tumor volume (>20 or < or = 20 cm (2), respectively) in the treatment of newly-diagnosed radiosurgery-ineligible. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 1351–1358. [Google Scholar] [PubMed]
- Lustig, R.A.; Seiferheld, W.; Berkey, B.; Yung, A.W.; Scarantino, C.; Movsas, B.; Jones, C.U.; Simpson, J.R.; Fishbach, J.; Curran, W.J., Jr. Imaging response in malignant glioma, RTOG 90-06. Am. J. Clin. Oncol. 2007, 30, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Werner-Wasik, M.; Scott, C.B.; Nelson, D.F.; Gaspar, L.E.; Murray, K.J.; Fischbach, J.A.; Nelson, J.S.; Weinstein, A.S.; Curran, W.J. Final report of a phase I/II trial of hyperfractionated and accelerated hyperfractionated radiation therapy with carmustine for adults with supratentorial malignant gliomas. Radiation Therapy Oncology Group Study 83-02. Cancer 1996, 77, 1535–1543. [Google Scholar] [CrossRef]
- Knisely, J.P.S.; Yue, N.; Chen, Z.; Nath, R.; Trumpore, S.; Duncan, J.S.; Studholme, C. Automatic three dimensional co-registration of diagnostic MRI and treatment planning CT for brain tumor radiotherapy treatment planning. Int. J. Radiat. Oncol. Biol. Phys. 1999. [Google Scholar] [CrossRef]
- Herman, M.G.; Pisansky, T.M.; Kruse, J.J.; Prisciandaro, J.I.; Davis, B.J.; King, B.F. Technical aspects of daily online positioning of the prostate for three-dimensional conformal radiotherapy using an electronic portal imaging device. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 1131–1140. [Google Scholar] [CrossRef]
- Guckenberger, M.; Sweeney, R.A.; Flickinger, J.C.; Gerszten, P.C.; Kersh, R.; Sheehan, J.; Sahgal, A. Clinical practice of image-guided spine radiosurgery––results from an international research consortium. Radiat. Oncol. 2011, 6, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.W.; Spratt, D.E.; Lovelock, M.; Bilsky, M.H.; Lis, E.; Ryu, S.; Sheehan, J.; Gerszten, P.C.; Chang, E.; Gibbs, I.; et al. International spine radiosurgery consortium consensus guidelines for target volume definition in spinal stereotactic radiosurgery. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Bowden, P.J.; See, A.W.; Dally, M.J.; Bittar, R.G. Stereotactic radiosurgery for brain and spine metastases. J. Clin. Neurosci. 2014, 21, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, S.; Detti, B.; Gadda, D.; Greto, D.; Furfaro, I.; Meacci, F.; Simontacchi, G.; Di Brina, L.; Bonomo, P.; Giacomelli, I.; et al. Organs at risk in the brain and their dose-constraints in adults and in children: A radiation oncologist’s guide for delineation in everyday practice. Radiother. Oncol. 2015, 114, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Van Baardwijk, A.; Bosmans, G.; Bentzen, S.M.; Boersma, L.; Dekker, A.; Wanders, R.; Wouters, B.G.; Lambin, P.; de Ruysscher, D. Radiation dose prescription for non-small-cell lung cancer according to normal tissue dose constraints: An in silico clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Carlson, D.J.; Brenner, D.J. The tumor radiobiology of SRS and SBRT: Are more than the 5 Rs involved? Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Okunieff, P.; Petersen, A.L.; Philip, A.; Milano, M.T.; Katz, A.W.; Boros, L.; Schell, M.C. Stereotactic Body Radiation Therapy (SBRT) for lung metastases. Acta Oncol. 2006, 45, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Kim, M.S.; Cho, L.C.; Dusenbery, K.; Sperduto, P.W. Radiobiological basis of SBRT and SRS. Int. J. Clin. Oncol. 2014, 19, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Shirato, H.; Nagata, Y.; Hiraoka, M.; Fujino, M.; Gomi, K.; Karasawa, K.; Hayakawa, K.; Niibe, Y.; Takai, Y.; et al. Stereotactic body radiotherapy (SBRT) for operable stage I non-small-cell lung cancer: Can SBRT be comparable to surgery? Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Wolff, D.; Stieler, F.; Welzel, G.; Lorenz, F.; Abo-Madyan, Y.; Mai, S.; Herskind, C.; Polednik, M.; Steil, V.; Wenz, F.; et al. Volumetric modulated arc therapy (VMAT) vs. serial tomotherapy, step-and-shoot IMRT and 3D-conformal RT for treatment of prostate cancer. Radiother. Oncol. 2009, 93, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.; Loewen, S.K.; Nichol, A.; Otto, K. 4D VMAT, gated VMAT, and 3D VMAT for stereotactic body radiation therapy in lung. Phys. Med. Biol. 2013, 58, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.M.; Popple, R.A.; Young, P.E.; Fiveash, J.B. Feasibility of single-isocenter volumetric modulated Arc radiosurgery for treatment of multiple brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, R.; Won, M.; Choucair, A.; Gillin, M.; Chakravarti, A.; Schultz, C.; Souhami, L.; Chen, A.; Pham, H.; Mehta, M. A phase II trial of accelerated radiotherapy using weekly stereotactic conformal boost for supratentorial glioblastoma multiforme: RTOG 0023. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.N.; Mehta, M.P.; Whelan, T.J.; Morris, D.E.; Hayman, J.A.; Flickinger, J.C.; Mills, M.; Rogers, C.L.; Souhami, L. The American Society for Therapeutic Radiology and Oncology (ASTRO) evidence-based review of the role of radiosurgery for malignant glioma. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Souhami, L.; Seiferheld, W.; Brachman, D.; Podgorsak, E.B.; Werner-Wasik, M.; Lustig, R.; Schultz, C.J.; Sause, W.; Okunieff, P.; Buckner, J.; et al. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: Report of Radiation Therapy Oncology Group 93-05 protocol. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Elaimy, A.L.; Mackay, A.R.; Lamoreaux, W.T.; Demakas, J.J.; Fairbanks, R.K.; Cooke, B.S.; Lamm, A.F.; Lee, C.M. Clinical outcomes of gamma knife radiosurgery in the salvage treatment of patients with recurrent high-grade glioma. World Neurosurg. 2013, 80, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.A.; Prados, M.; Lamborn, K.R.; Smith, V.; Sneed, P.K.; Chang, S.; Nicholas, K.M.; Wara, W.M.; Devriendt, D.; Kunwar, S.; et al. Phase II study of high central dose Gamma Knife radiosurgery and marimastat in patients with recurrent malignant glioma. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1397–1404. [Google Scholar] [CrossRef]
- Murphy, E.S.; Chao, S.T.; Angelov, L.; Vogelbaum, M.A.; Barnett, G.; Jung, E.; Recinos, V.R.; Mohammadi, A.; Suh, J.H. Radiosurgery for Pediatric Brain Tumors. Pediatr. Blood Cancer 2016, 63, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Kondziolka, D.; Flickinger, J.; Lunsford, L.D. Radiosurgery for brain metastases. Prog. Neurol. Surg. 2012, 25, 115–122. [Google Scholar] [PubMed]
- Minniti, G.; Amichetti, M.; Enrici, R.M. Radiotherapy and radiosurgery for benign skull base meningiomas. Radiat. Oncol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Kocher, M.; Wittig, A.; Piroth, M.D.; Treuer, H.; Seegenschmiedt, H.; Ruge, M.; Grosu, A.L.; Guckenberger, M. Stereotactic radiosurgery for treatment of brain metastases. A report of the DEGRO Working Group on Stereotactic Radiotherapy. Strahlenther. Onkol. 2014, 190, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, H.; Shirato, H.; Onimaru, R.; Kagei, K.; Ikeda, J.; Ishii, N.; Sawamura, Y.; Miyasaka, K. Hypofractionated stereotactic radiotherapy alone without whole-brain irradiation for patients with solitary and oligo brain metastasis using noninvasive fixation of the skull. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 793–800. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.D.; Bentzen, S.M.; Harari, P.M. Biologic basis for combining drugs with radiation. Semin. Radiat. Oncol. 2006, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Steel, G.G. Terminology in the description of drug-radiation interactions. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1145–1150. [Google Scholar] [CrossRef]
- Steel, G.G.; Peckham, M.J. Exploitable mechanisms in combined radiotherapy-chemotherapy: The concept of additivity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 85–91. [Google Scholar] [CrossRef]
- Brandes, A.A.; Bartolotti, M.; Marucci, G.; Ghimenton, C.; Agati, R.; Fioravanti, A.; Mascarin, M.; Volpin, L.; Ammannati, F.; Masotto, B.; et al. New perspectives in the treatment of adult medulloblastoma in the era of molecular oncology. Crit. Rev. Oncol. Hematol. 2015, 94, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, K.; Spiegler, B.J.; Fung, S.; Panzarella, T.; Mabbott, D.J.; Jewitt, N.; D’Agostino, N.M.; Mason, W.P.; Bouffet, E.; Tabori, U.; et al. Early aging in adult survivors of childhood medulloblastoma: Long-term neurocognitive, functional, and physical outcomes. Neuro-oncology 2011, 13, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.S.; Blackledge, G.R.; Slack, J.A.; Rustin, G.J.; Smith, D.B.; Stuart, N.S.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.; Brampton, M.H. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar] [PubMed]
- Bower, M.; Newlands, E.S.; Bleehen, N.M.; Brada, M.; Begent, R.J.H.; Calvert, H.; Colquhoun, I.; Lewis, P.; Brampton, M.H. Multicentre CRC phase II trial of temozolomide in recurrent or progressive high-grade glioma. Cancer Chemother. Pharmacol. 1997, 40, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Khosla, D. Concurrent therapy to enhance radiotherapeutic outcomes in glioblastoma. Ann. Transl. Med. 2016, 4, 2–9. [Google Scholar]
- He, Z.; Ping, Y.; Xu, S.; Lin, Y.; Yu, S.; Kung, H.; Bian, X. Lower MGMT expression predicts better prognosis in proneural-like glioblastoma. Int. J. Exp. Med. 2015, 8, 20287–20294. [Google Scholar]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Dewire, M.; Ryall, S.; Buczkowicz, P.; Leach, J.; Miles, L.; Ramani, A.; Brudno, M.; Kumar, S.S.; Drissi, R.; et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: Implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol. Commun. 2016. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.M.; Combi, R.; Cajola, L.; Patrizi, A.; Redaelli, S.; Bentivegna, A.; Baronchelli, S.; Maira, G.; Pollo, B.; Mangiola, A.; et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene 2009, 28, 1807–1811. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.M.; Colman, S.; Potter, N.E.; van Delft, F.W.; Lillis, S.; Carnicer, M.J.; Kearney, L.; Watts, C.; Greaves, M. Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Reports 2015, 4, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Rath, B.H.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. Microenvironmental regulation of glioblastoma radioresponse. Clin. Cancer Res. 2010, 16, 6049–6059. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Lubelski, D.; Miller, T.E.; Rich, J.N. Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol. Aspects Med. 2014, 39, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Herold-mende, C.; Mock, A. Microenvironment and brain tumor stem cell maintenance: Impact of the niche. Anticancer Agents Med. Chem. 2014, 14, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Holland, E.C. Brain tumor treatment increases the number of cancer stem-like cells. Expert Rev. Neurother. 2009, 9, 1447–1449. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol. 2014, 234, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Fang, B. Development of synthetic lethality anticancer therapeutics. J. Med. Chem. 2014, 57, 7859–7873. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.J.; Mulligan, E.A.; Grogan, P.T.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Curtin, N.J.; Lou, Z.; Decker, P.A.; Wu, W.; et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol. Cancer Ther. 2009, 8, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Jena, R.; Burnet, N.G.; Meira, L.B.; Jeynes, J.C.G.; Kirkby, K.J.; Kirkby, N.F. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat. Oncol. 2013, 8, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. 2015, 5749, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.A.; Callow, M.G.; Waldbaum, L.; Thomas, S.; Ruggieri, R. MRK, a mixed lineage kinase-related molecule that plays a role in gamma-radiation-induced cell cycle arrest. J. Biol. Chem. 2002, 277, 13873–13882. [Google Scholar] [CrossRef] [PubMed]
- Tosti, E.; Waldbaum, L.; Warshaw, G.; Gross, E.A.; Ruggieri, R. The stress kinase MRK contributes to regulation of DNA damage checkpoints through a p38γ-independent pathway. J. Biol. Chem. 2004, 279, 47652–47660. [Google Scholar] [CrossRef] [PubMed]
- Vanan, I.; Dong, Z.; Tosti, E.; Warshaw, G.; Symons, M.; Ruggieri, R. Role of a DNA Damage Checkpoint Pathway in Ionizing Radiation-Induced Glioblastoma Cell Migration and Invasion. Cell. Mol. Neurobiol. 2012, 32, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, R.; The Feinstein Institute, Manhasset, NY, USA. Unpublished data. 2016.
- D’Avenia, P.; Porrello, A.; Berardo, M.; Angelo, M.D.; Soddu, S.; Arcangeli, G.; Sacchi, A.; D’Orazi, G. Tp53-gene transfer induces hypersensitivity to low doses of X-rays in glioblastoma cells: A strategy to convert a radio-resistant phenotype into a radiosensitive one. Cancer Lett. 2006, 231, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.K.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. CDK1-dependent regulation of the mitotic inhibitor Wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.E.; de Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Niers, J.M.; van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; Peter Vandertop, W.; et al. In Silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma shahryar. Cancer Cell 2011, 18, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.L.; Noske, D.P.; et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol. Cancer Ther. 2013, 12, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A. Editorial on “targeting Wee1 for the treatment of pediatric high-grade gliomas”. Neuro. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; Demuth, T.; Chinnaiyan, P. Targeting Radiation-Induced G2 Checkpoint Activation with the Wee-1 Inhibitor MK-1775 in Glioblastoma Cell Lines. Mol. Cancer Ther. 2011, 10, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Rodemann, H.P.; Dittmann, K.; Toulany, M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int. J. Radiat. Biol. 2007, 83, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Carrera Nuclear phosphoinositide 3-kinase β controls doublestrand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Hitomi, M.; Venere, M.; Flavahan, W.A.; Yang, K.; Kim, Y.; Minhas, S.; Rich, J.N.; Hjelmeland, A.B. Glioma stem cell maintenance: The role of the microenvironment. Curr. Pharm. Des. 2011, 17, 2386–2401. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.S.; Didier, S.; Murray, D.W.; Turner, T.H.; Issaivanan, M.; Ruggieri, R.; Al-Abed, Y.; Symons, M. Semapimod sensitizes glioblastoma tumors to ionizing radiation by targeting microglia. PLoS ONE 2014, 9, e95885. [Google Scholar] [CrossRef] [PubMed]
- Knisely, J.P.; Rockwell, S. Importance of hypoxia in the biology and treatment of brain tumors. Neuroimaging Clin. N. Am. 2002, 12, 525–536. [Google Scholar] [CrossRef]
- Fulton, D.S.; Urtasun, R.C.; Shin, K.H.; Geggie, P.H.; Thomas, H.; Muller, P.J.; Moody, J.; Tanasichuk, H.; Mielke, B.; Johnson, E. Misonidazole combined with hyperfractionation in the management of malignant glioma. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 1709–1712. [Google Scholar] [CrossRef]
- Collingridge, D.R.; Piepmeier, J.M.; Rockwell, S.; Knisely, J.P. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother. Oncol. 1999, 53, 127–131. [Google Scholar] [CrossRef]
- Yasui, H.; Asanuma, T.; Kino, J.; Yamamori, T.; Meike, S.; Nagane, M.; Kubota, N.; Kuwabara, M.; Inanami, O. The prospective application of a hypoxic radiosensitizer, doranidazole to rat intracranial glioblastoma with blood brain barrier disruption. BMC Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Kohshi, K.; Beppu, T.; Tanaka, K.; Ogawa, K.; Inoue, O.; Kukita, I.; Clarke, R.E. Potential roles of hyperbaric oxygenation in the treatments of brain tumors. Undersea Hyperb. Med. 2013, 40, 351–362. [Google Scholar] [PubMed]
- Dowling, S.; Fischer, J.J.; Rockwell, S. Fluosol and hyperbaric oxygen as an adjunct to radiation therapy in the treatment of malignant gliomas: A pilot study. Biomater. Artif. Cells Immobilization Biotechnol. 1992, 20, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J. Hypoxic radiosensitization: Adored and ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef] [PubMed]
- Halperin, E.C.; Herndon, J.; Schold, S.C.; Brown, M.; Vick, N.; Cairncross, J.G.; Macdonald, D.R.; Gaspar, L.; Fischer, B.; Dropcho, E.; et al. A phase III randomized prospective trial of external beam radiotherapy, mitomycin C, carmustine, and 6-mercaptopurine for the treatment of adults with anaplastic glioma of the brain. CNS Cancer Consortium. Int. J. Radiat. Oncol. Biol. Phys. 1996, 34, 793–802. [Google Scholar] [CrossRef]
- Del Rowe, J.; Scott, C.; Werner-Wasik, M.; Bahary, J.P.; Curran, W.J.; Urtasun, R.C.; Fisher, B. Single-arm, open-label phase II study of intravenously administered tirapazamine and radiation therapy for glioblastoma multiforme. J. Clin. Oncol. 2000, 18, 1254–1259. [Google Scholar] [PubMed]
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg. Focus 2014. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; Sloan, A.E.; Iliopoulos, O.; Hjelmeland, A.B.; Rich, J.N. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012, 19, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X.G. The canonical notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Yoon, K.; Dang, L.; Tokunaga, A.; Gaiano, N. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 2007, 449, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [PubMed]
- Teodorczyk, M.; Schmidt, M.H. Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Chen, J.; Kesari, S.; Rooney, C.; Strack, P.R.; Chen, J.; Shen, H.; Wu, L.; Griffin, J.D. Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 2010, 1, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C. Genomic profiles of glioma. Curr. Neurol. Neurosci. Rep. 2011, 11, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, S.O.; Jin, X.; Ham, S.W.; Kim, J.; Park, J.B.; Lee, J.Y.; Kim, S.C.; Kim, H. Epidermal growth factor receptor variant III renders glioma cancer cells less differentiated by JAGGED1. Tumor Biol. 2014, 36, 2921–2928. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Latha, J.D.; Hjelmeland, A.B.; White, R.R.; Rich, J.N.; Sullenger, B. A notch promotes radioresistance of glioma stem cells. Stem Cells 2011, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/Beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, K.H.; Lee, H.; Yang, H.; Kim, D.; Kang, W.; Jin, J.; Joo, K.M.; Lee, J.; Nam, D.-H. Wnt activation is implicated in glioblastoma radioresistance. Lab. Investig. 2012, 92, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.H.; Zhang, H.; Xiao, Y.; Perry, S.R.; et al. PLAGL2 Regulates Wnt Signaling to Impede Differentiation in Neural Stem Cells and Gliomas. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Wild-Bode, C.; Platten, M.; Grimmel, C.; Melkonyan, H.S.; Dichgans, J.; Weller, M. Secreted Frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene 2000, 19, 4210–4220. [Google Scholar] [CrossRef] [PubMed]
- Götze, S.; Wolter, M.; Reifenberger, G.; Müller, O.; Sievers, S. Frequent promoter hypermethylation of Wnt pathway inhibitor genes in malignant astrocytic gliomas. Int. J. Cancer 2010, 126, 2584–2593. [Google Scholar] [PubMed]
- Rheinbay, E.; Suvà, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Mao, X.; Wang, P.; Liu, B.; Zhang, X.; Jiang, X.; Zhong, C.; Huo, J.; Jin, J.; Zhuo, Y. The expression profile of FRAT1 in human gliomas. Brain Res. 2010, 1320, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Kuai, D.; Cai, S.; Xue, N.; Liu, Y.; Hao, J.; Fan, Y.; Jin, J.; Mao, X.; Liu, B.; et al. Knockdown of FRAT1 Expression by RNA Interference Inhibits Human Glioblastoma Cell Growth, Migration and Invasion. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Chen, Y.Y.; Li, B.A.; Tan, G.W.; Liu, X.Y.; Shen, S.H.; Zhu, H.W.; Wang, H.D. Decreased pygopus 2 expression suppresses glioblastoma U251 cell growth. J. Neurooncol. 2010, 100, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Xu, L.; Chen, Y.; Yan, H.; Hazawa, M.; Doan, N.; Said, J.W.; Ding, L.W.; Liu, L.Z.; Yang, H.; et al. Genomic and Functional Analysis of the E3 Ligase PARK2 in Glioma. Cancer Res. 2015, 75, 1815–1828. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Salaroli, R.; Ronchi, A.; Buttarelli, F.R.; Cortesi, F.; Marchese, V.; Della Bella, E.; Renna, C.; Baldi, C.; Giangaspero, F.; Cenacchi, G. Wnt activation affects proliferation, invasiveness and radiosensitivity in medulloblastoma. J. Neurooncol. 2015, 121, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Angot, E.; Ruat, M. Sonic Hedgehog signaling in the mammalian brain. J. Neurochem. 2010, 113, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Rey, J.A.; Castresana, J.S. The sonic hedgehog-GLI1 signaling pathway in brain tumor development. Expert Opin. Ther. Targets 2012, 12, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Palma, V.; Dahmane, N. Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002, 3, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.; Ellsworth, R.; Hsieh, D. Hedgehog/GLI1 regulates IGF dependent malignant behaviors in glioma stem cells. J. Cell. Physiol. 2011, 226, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Lorente, A.; Castresana, J.S. Hedgehog signalling in medulloblastoma, glioblastoma and neuroblastoma. Oncol. Rep. 2008, 19, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Michael, L.E.; Westerman, B.A.; Ermilov, A.; Wang, A.; Ferris, J.; Liu, J.; Blom, M.; Ellison, D.; van Lohuizen, M.; Dlugosz, A. Bmi1 Is Required for Hedgehog Pathway––Driven medulloblastome expansion. Neoplasia 2008, 10, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zencak, D.; Lingbeek, M.; Kostic, C.; Tekaya, M.; Tanger, E.; Hornfeld, D.; Jaquet, M.; Munier, F.L.; Schorderet, D.F.; van Lohuizen, M.; et al. Bmi1 loss produces an increase in astroglial cells and a decrease in neural stem cell population and proliferation. J. Neurosci. 2005, 25, 5774–5783. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. A treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Haley, E.M.; Kim, Y. The role of basic fibroblast growth factor in glioblastoma multiforme and glioblastoma stem cells and in their in vitro culture. Cancer Lett. 2014, 346, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Hitomi, M.; Gallagher, J.; Gadani, S.P.; Adkins, J.; Vasanji, A.; Liu, L.; Eyler, C.E.; Heddleston, J.M.; Wu, Q.; et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011. [Google Scholar] [CrossRef] [PubMed]
- Fuks, Z.; Persaud, R.S.; Alfieri, A.; McLoughlin, M.; Ehleiter, D.; Schwartz, J.L.; Seddon, A.P.; Cordon-Cardo, C.; Haimovitz-Friedman, A. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res. 1994, 54, 2582–2590. [Google Scholar] [PubMed]
- Haimovitz-Friedman, A.; Balaban, N.; McLoughlin, M.; Ehleiter, D.; Michaeli, J.; Vlodavsky, I.; Fuks, Z. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 1994, 54, 2591–2597. [Google Scholar] [PubMed]
- Ader, I.; Toulas, C.; Dalenc, F.; Delmas, C.; Bonnet, J.; Cohen-Jonathan, E.; Favre, G. RhoB controls the 24 kDa FGF-2-induced radioresistance in HeLa cells by preventing post-mitotic cell death. Oncogene 2002, 21, 5998–6006. [Google Scholar] [CrossRef] [PubMed]
- Delmas, C.; Heliez, C.; Cohen-Jonathan, E.; End, D.; Bonnet, J.; Favre, G.; Toulas, C. Farnesyltransferase inhibitor, R115777, reverses the resistance of human glioma cell lines to ionizing radiation. Int. J. Cancer 2002, 100, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Srougi, M.C.; Burridge, K. The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Monferran, S.; Skuli, N.; Delmas, C.; Favre, G.; Bonnet, J.; Cohen-Jonathan-Moyal, E.; Toulas, C. AlphavBeta3 and AlphavBeta5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int. J. Cancer 2008, 123, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mamouni, K.; Cristini, A.; Guirouilh-Barbat, J.; Monferran, S.; Lemarie, A.; Faye, J.C.; Lopez, B.S.; Favre, G.; Sordet, O. RhoB promotes gammaH2AX dephosphorylation and DNA double-strand break repair. Mol. Cell Biol. 2014, 34, 3144–3155. [Google Scholar] [CrossRef] [PubMed]
- Ducassou, A.; Uro-Coste, E.; Verrelle, P.; Filleron, T.; Benouaich-Amiel, A.; Lubrano, V.; Sol, J.C.; Delisle, M.B.; Favre, G.; Ken, S.; et al. αvβ3 Integrin and Fibroblast growth factor receptor 1 (FGFR1): Prognostic factors in a phase I-II clinical trial associating continuous administration of Tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur. J. Cancer 2013, 49, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Ader, I.; Delmas, C.; Skuli, N.; Bonnet, J.; Schaeffer, P.; Bono, F.; Cohen-Jonathan-Moyal, E.; Toulas, C. Preclinical evidence that SSR128129E––A novel small-molecule multi-fibroblast growth factor receptor blocker––Radiosensitises human glioblastoma. Eur. J. Cancer 2014, 50, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Burrows, R.C.; Wancio, D.; Levitt, P.; Lillien, L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 1997, 19, 251–267. [Google Scholar] [CrossRef]
- Chakravarti, A.; Dicker, A.; Mehta, M. The contribution of epidermal growth factor receptor (EGFR) signaling pathway to radioresistance in human gliomas: A review of preclinical and correlative clinical data. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Zalutsky, M.R.; Fuller, G.N.; Archer, G.E.; Friedman, H.S.; Kwatra, M.M.; Bigner, S.H.; Bigner, D.D. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA 1990, 87, 4207–4211. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Chakladar, A.; Delaney, M.A.; Latham, D.E.; Loeffler, J.S. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002, 62, 4307–4315. [Google Scholar] [PubMed]
- Lammering, G.; Hewit, T.H.; Hawkins, W.T.; Contessa, J.N.; Reardon, D.B.; Lin, P.S.; Valerie, K.; Dent, P.; Mikkelsen, R.B.; Schmidt-Ullrich, R.K. Epidermal growth factor receptor as a genetic therapy target for carcinoma cell radiosensitization. J. Natl. Cancer Inst. 2004, 279, 15130–15141. [Google Scholar] [CrossRef]
- Lammering, G.; Lin, P.S.; Contessa, J.N.; Hampton, J.L.; Valerie, K.; Schmidt-Ullrich, R.K. Adenovirus-mediated overexpression of dominant negative epidermal growth factor receptor-CD533 as a gene therapeutic approach radiosensitizes human carcinoma and malignant glioma cells. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 775–784. [Google Scholar] [CrossRef]
- Lammering, G.; Valerie, K.; Lin, P.S.; Mikkelsen, R.B.; Contessa, J.N.; Feden, J.P.; Farnsworth, J.; Dent, P.; Schmidt-Ullrich, R.K. Radiosensitization of malignant glioma cells through overexpression of dominant-negative epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 682–690. [Google Scholar] [PubMed]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A Phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liffers, K.; Lamszus, K.; Schulte, A. EGFR amplification and glioblastoma stem-like cells. Stem Cells Int. 2015, 2015, 427518–427529. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Achim, C.L.; Katyal, S.; Wiley, C.A.; Shiratori, M.; Wang, G.; Oshika, E.; Petersen, B.E.; Li, J.M.; Michalopoulos, G.K. Expression of HGF and cMET in the developing and adult brain. Dev. Brain Res. 1997, 102, 299–303. [Google Scholar] [CrossRef]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; vande Woude, G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar] [PubMed]
- Moriyama, T.; Kataoka, H.; Koono, M.; Wakisaka, S. Expression of hepatocyte growth factor/scatter factor and its receptor c-Met in brain tumors: Evidence for a role in progression of astrocytic tumors. Int. J. Mol. Med. 1999, 3, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Bowers, D.C.; Fan, S.; Walter, K.A.; Abounader, R.; Williams, J.A.; Rosen, E.M.; Laterra, J. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res. 2000, 60, 4277–4283. [Google Scholar] [PubMed]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-oncology 2005, 7, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-Met pathway potentiates glioblastoma responses to γ-radiation. Clin. Cancer Res. 2005, 11, 4479–4486. [Google Scholar] [CrossRef] [PubMed]
- Matheny, R.W., Jr.; Adamo, M.L. Current perspectives on Akt Akt-ivation and Akt-ions. Exp. Biol. Med. (Maywood). 2009, 234, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Matsutani, T.; Nagai, Y.; Mine, S.; Murai, H.; Saeki, N.; Iwadate, Y. Akt/protein kinase B overexpression as an accurate prognostic marker in adult diffuse astrocytoma. Acta Neurochir. (Wien). 2009, 151, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Trampe-Kieslich, A.; Reifenberger, G. Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol. Appl. Neurobiol. 2005, 31, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Reifenberger, J.; Blaschke, B.; Reifenberger, G. Hypermethylation and transcriptional downregulation of the carboxyl-terminal modulator protein gene in glioblastomas. J. Natl. Cancer Inst. 2004, 96, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Squatrito, M.; Carbajal, E.; Holland, E.C. Glioma formation, cancer stem cells, and Akt signaling. Stem Cell Rev. 2008, 4, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gallia, G.L.; Tyler, B.M.; Hann, C.L.; Siu, I.M.; Giranda, V.L.; Vescovi, A.L.; Brem, H.; Riggins, G.J. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol. Cancer Ther. 2009, 8, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Foo, W.; Lafiura, K.M.; Mclendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2009, 26, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Włodarski, P.; Grajkowska, W.; Łojek, M.; Rainko, K.; Jóźwiak, J. Activation of Akt and Erk pathways in medulloblastoma. Folia Neuropathol. 2006, 44, 214–220. [Google Scholar] [PubMed]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Kim, J.S.; Waldman, T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Khan, A.; Danish, S.; Haffty, B.G.; Sabaawy, H.E. Radiosensitization of primary human glioblastoma stem-like cells with low-dose AKT Inhibition. Mol. Cancer Ther. 2015, 14, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2013, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Haas-Kogan, D.A. The combination of novel targeted molecular agents and radiation in the treatment of pediatric gliomas. Front. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Qin, Z.; Liang, Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim. Biophys. Sin. 2009, 41, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Cerniglia, G.J.; Karar, J.; Koumenis, C. Inhibition of autophagy as a strategy to augment radiosensitization by the dual PI3K/mTOR inhibitor NVP-BEZ235. Int. J. Radiat. Oncol. 2012, 82, 1230–1240. [Google Scholar] [CrossRef]
- Wang, W.; Long, L.; Yang, N.; Zhang, Q.; Ji, W.; Zhao, J.; Qin, Z.; Wang, Z.; Chen, G.; Liang, Z. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; DeCarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Lee, Y.S.; Lee, M.; Lee, H.J.; Lee, B.W.; Lee, H.J.; Lee, K.E.; Jung, M.K.; et al. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res. 2013, 73, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; Dubois, L.; Schaaf, M.B.; van den Beucken, T.; Lieuwes, N.; Keulers, T.G.; Savelkouls, K.G.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; et al. Deregulation of cap-dependent mRNA translation increases tumour radiosensitivity through reduction of the hypoxic fraction. Radiother. Oncol. 2011, 99, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar] [PubMed]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; Jaeckle, K.A.; Buckner, J.C. North central cancer treatment group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Dietz, A.B.; Kaufmann, T.J.; Gustafson, M.P.; Brown, P.D.; Uhm, J.H.; Rao, R.D.; Doyle, L.; et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin. Cancer Res. 2010, 16, 5573–5580. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Cho, B.J.; Lee, D.J.; Hwang, Y.H.; Chun, S.H.; Kim, H.H.; Kim, I.A. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014, 14, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-oncology 2011, 13, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Gil del Alcazar, C.R.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR Inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Tini, P.; Toscano, M.; Allavena, G.; Angeletti, F.; Manai, F.; Miracco, C.; Comincini, S.; Pirtoli, L. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. J. Cell. Physiol. 2014, 229, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ezhilarasan, R.; Phillips, E.; Gallego-Perez, D.; Sparks, A.; Taylor, D.; Ladner, K.; Furuta, T.; Sabit, H.; Chhipa, R.; et al. Serine/Threonine kinase MLK4 determines mesenchymal identity in glioma stem cells in an NF-kappaB-dependent manner. Cancer Cell 2016, 29, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ghildiyal, R.; Mehta, V.S.; Sen, E. ATM-NFκB axis-driven TIGAR regulates sensitivity of glioma cells to radiomimetics in the presence of TNFα. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hein, A.; Ouellette, M.; Yan, Y. Radiation-induced signaling pathways that promote cancer cell survival (Review). Int. J. Oncol. 2014, 45, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.X.; Liu, R.Y.; Wu, C.M.; Zhao, Y.S.; Li, Y.; Yao, Y.Q.; Xu, Y.H. DNA damage-induced NF-κB activation in human glioblastoma cells promotes miR-181b expression and cell proliferation. Cell. Physiol. Biochem. 2015, 35, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Séhédic, D.; Cikankowitz, A.; Hindré, F.; Davodeau, F.; Garcion, E. Nanomedicine to overcome radioresistance in glioblastoma stem-like cells and surviving clones. Trends Pharmacol. Sci. 2015, 36, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Zhu, X.; Moore, M.J.; Lu, L.; Yaszemski, M.J.; Windebank, A.J. Effects of NF-Kappa-B decoy oligonucleotides released from biodegradable polymer microparticles on a glioblastoma cell line. Biomaterials 2002, 23, 2773–2781. [Google Scholar] [CrossRef]
- Brassesco, M.S.; Roberto, G.M.; Morales, A.G.; Oliveira, J.C.; Delsin, L.E.; Pezuk, J.A.; Valera, E.T.; Carlotti, C.G.; Rego, E.M.; de Oliveira, H.F.; et al. Inhibition of NF-κB by dehydroxymethylepoxyquinomicin suppresses invasion and synergistically potentiates temozolomide and γ-radiation cytotoxicity in glioblastoma cells. Chemother. Res. Pract. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.P.; Nozell, S.E.; Benveniste, E.T. NF-kappaB and STAT3 signaling in glioma: Targets for future therapies. Expert Rev. Neurother. 2010, 10, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 1471–2407. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Miyake, S.; Nakanowatari, S. Signaling pathways in glioblastoma cancer stem cells: A role of Stat3 as a potential therapeutic target. Austin J. Cancer Clin. Res. 2015, 2, 1030–1035. [Google Scholar]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. STATs: Signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Swiatek-Machado, K.; Kaminska, B. STAT signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 189–208. [Google Scholar] [PubMed]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive activation of Stat3α in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O. Aberrant Stat3 signaling by interleukin-4 in malignant glioma cells: Involvement of IL-13Rα2. Cancer Res. 2005, 65, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Magrassi, L.; De-Fraja, C.; Conti, L.; Di Gennaro, I.; Butti, G.; Govoni, S. Variations in the levels of the JAK/STAT and ShcA proteins in human brain tumors. Anticancer Res. 1998, 18, 2381–2387. [Google Scholar] [PubMed]
- Ouédraogo, Z.G.; Müller-Barthélémy, M.; Kemeny, J.L.; Dedieu, V.; Biau, J.; Khalil, T.; Raoelfils, L.I.; Granzotto, A.; Pereira, B.; Beaudoin, C.; et al. STAT3 serine 727 phosphorylation: A relevant target to radiosensitize human glioblastoma. Brain Pathol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Deangelis, S.; Foust, E.; Fuchs, J.; Li, C.; Li, P.K.; Schwartz, E.B.; Lesinski, G.B.; Benson, D.; Lü, J.; et al. A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol. Cancer 2010, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Chang, Y.L.; Huang, P.I.; Chiou, G.Y.; Tseng, L.M.; Chiou, S.H.; Chen, M.H.; Chen, M.T.; Shih, Y.H.; Chang, C.H.; et al. Resveratrol suppresses tumorigenicity and enhances radiosensitivity in primary glioblastoma tumor initiating cells by inhibiting the STAT3 axis. J. Cell. Physiol. 2012, 227, 976–993. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelley, K.; Knisely, J.; Symons, M.; Ruggieri, R. Radioresistance of Brain Tumors. Cancers 2016, 8, 42. https://doi.org/10.3390/cancers8040042
Kelley K, Knisely J, Symons M, Ruggieri R. Radioresistance of Brain Tumors. Cancers. 2016; 8(4):42. https://doi.org/10.3390/cancers8040042
Chicago/Turabian StyleKelley, Kevin, Jonathan Knisely, Marc Symons, and Rosamaria Ruggieri. 2016. "Radioresistance of Brain Tumors" Cancers 8, no. 4: 42. https://doi.org/10.3390/cancers8040042
APA StyleKelley, K., Knisely, J., Symons, M., & Ruggieri, R. (2016). Radioresistance of Brain Tumors. Cancers, 8(4), 42. https://doi.org/10.3390/cancers8040042