
HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies


Abstract
:1. Epidemiology and Etiology of HNSCC
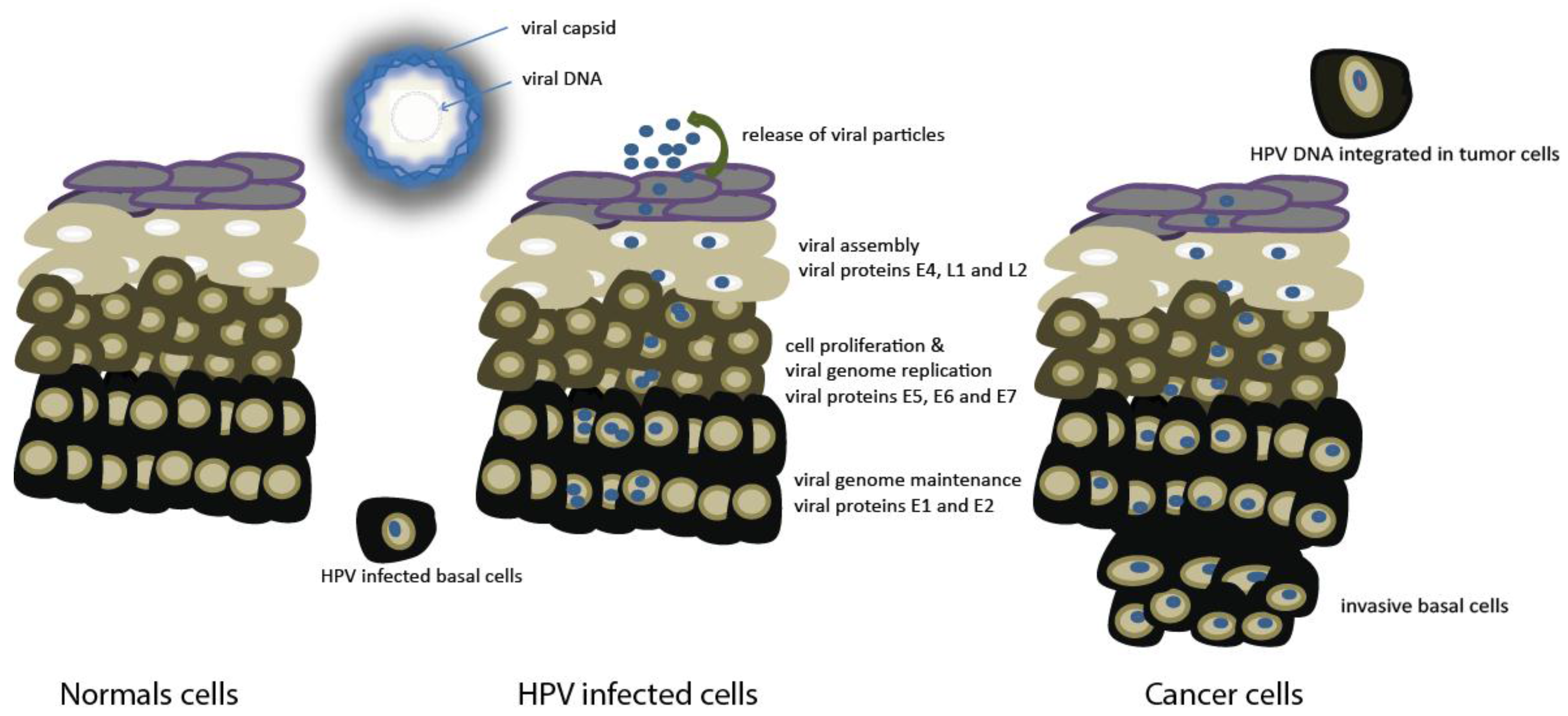
2. Molecular Pathogenesis of HPV Positive HNSCC
3. Treatment Response
4. Biological Basis for the Treatment Response
5. Biological Markers in HNSCC
6. Possibilities to Increase the Current Therapeutic Window
6.1. De-Intensification of Current Therapy Options
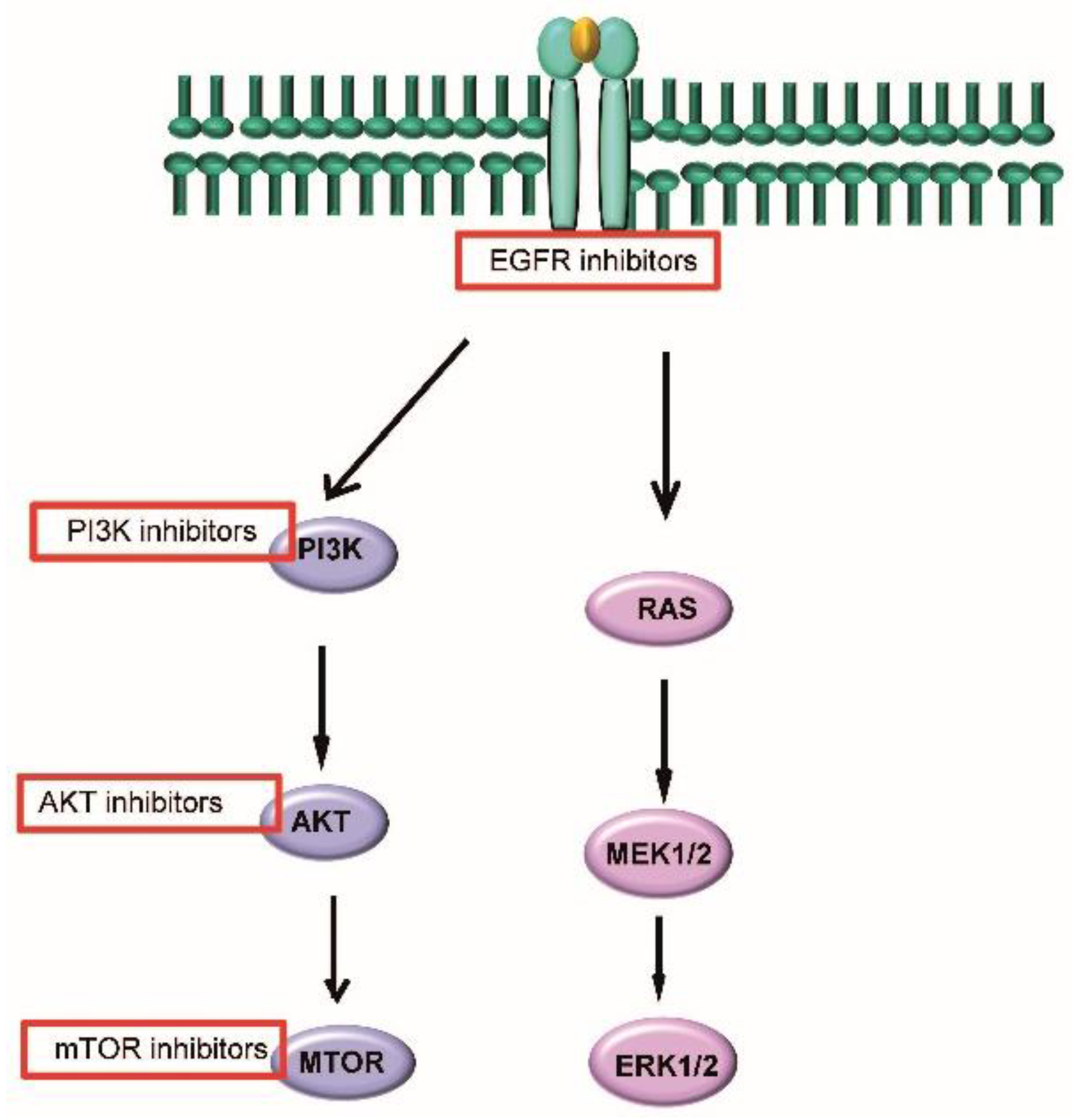
6.2. Targeted Molecular Agents
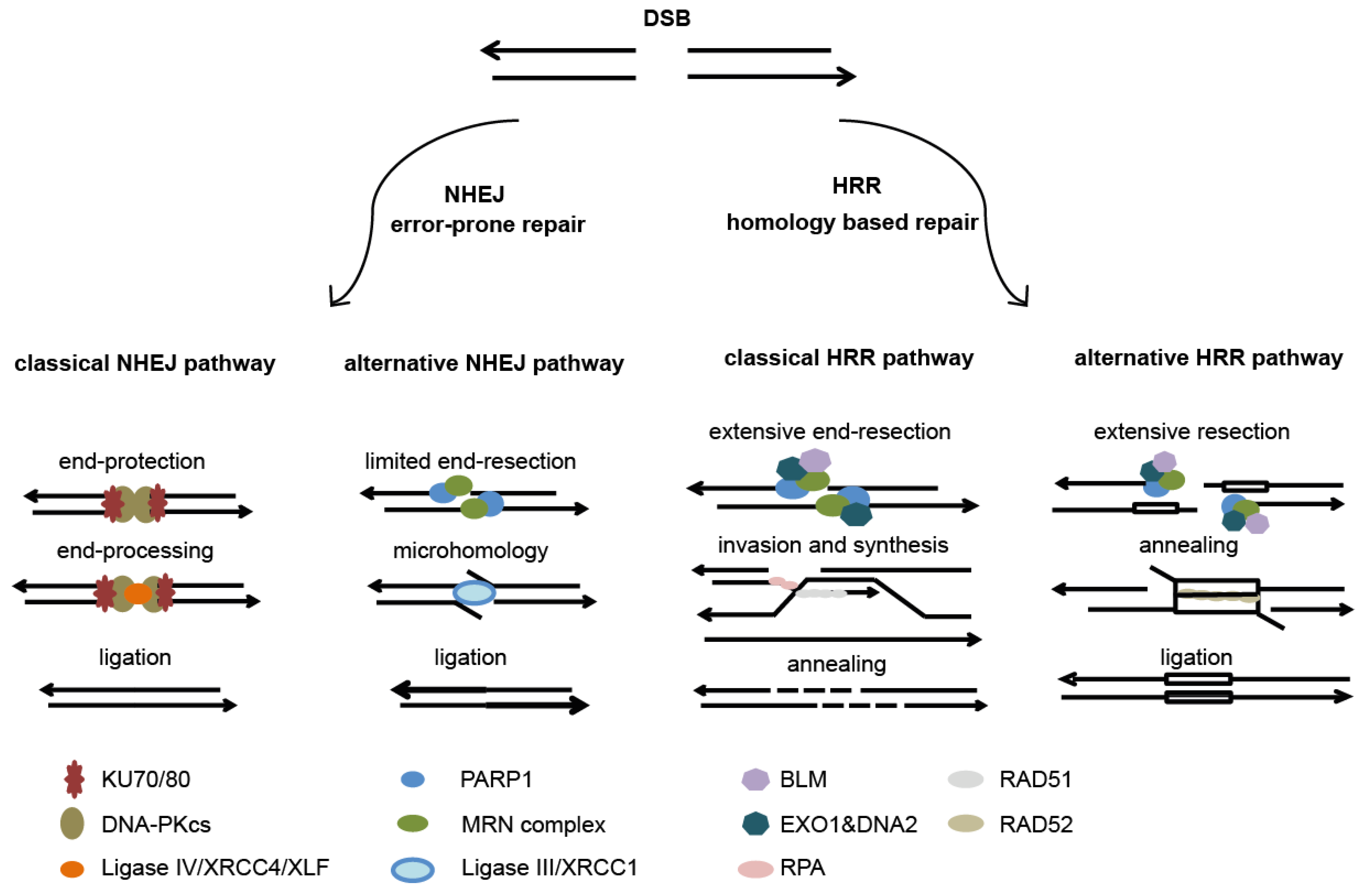
6.3. Summary of DNA Damage Repair Mechanisms
6.4. Targeting DNA Repair by Modulating Radiotherapy Response
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Investig. 2012, 122, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Andre, K.; Schraub, S.; Mercier, M.; Bontemps, P. Role of alcohol and tobacco in the aetiology of head and neck cancer: A case-control study in the Doubs region of France. Eur. J. Cancer B Oral Oncol. 1995, 31, 301–309. [Google Scholar] [CrossRef]
- Kang, H.; Kiess, A.; Chung, C.H. Emerging biomarkers in head and neck cancer in the era of genomics. Nat. Rev. Clin. Oncol. 2015, 12, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Takayama, K.; Choi, B.C.; Pak, A.W. A nested case-control study on alcohol drinking, tobacco smoking, and cancer. Cancer Detect. Prev. 1996, 20, 557–565. [Google Scholar] [PubMed]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; Forastiere, A.A. Head and neck cancer: Changing epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2008, 83, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Sturgis, E.M.; Cinciripini, P.M. Trends in head and neck cancer incidence in relation to smoking prevalence: An emerging epidemic of human papillomavirus-associated cancers? Cancer 2007, 110, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Agalliu, I.; Gapstur, S.; Chen, Z.; Wang, T.; Anderson, R.L.; Teras, L.; Kreimer, A.R.; Hayes, R.B.; Freedman, N.D.; Burk, R.D. Associations of Oral alpha-, beta-, and gamma-human papillomavirus types with risk of incident head and neck cancer. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, G.C.; Smith, M.A.; Harris, S.L.; Kimple, R.J. Review of the clinical and biologic aspects of human papillomavirus-positive squamous cell carcinomas of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Castellsague, X.; Alemany, L.; Quer, M.; Halec, G.; Quiros, B.; Tous, S.; Clavero, O.; Alos, L.; Biegner, T.; Szafarowski, T.; et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. J. Natl. Cancer Inst. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P.; Primdahl, H.; Johansen, J.; Kristensen, C.A.; Andersen, E.; Andersen, L.J.; Evensen, J.F.; Eriksen, J.G.; Overgaard, J. Impact of HPV-associated p16-expression on radiotherapy outcome in advanced oropharynx and non-oropharynx cancer. Radiother. Oncol. 2014, 113, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Ang, M.K.; Yin, X.Y.; Patel, M.R.; Fritchie, K.; Thorne, L.; Muldrew, K.L.; Hayward, M.C.; Sun, W.; Wilkerson, M.D.; et al. Different cellular p16(INK4a) localisation may signal different survival outcomes in head and neck cancer. Br. J. Cancer 2012, 107, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Zhang, Q.; Kong, C.S.; Harris, J.; Fertig, E.J.; Harari, P.M.; Wang, D.; Redmond, K.P.; Shenouda, G.; Trotti, A.; et al. p16 protein expression and human papillomavirus status as prognostic biomarkers of nonoropharyngeal head and neck squamous cell carcinoma. J. Clin. Oncol. 2014, 32, 3930–3938. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Castellsague, X.; Chaturvedi, A.; Goodman, M.T.; Snijders, P.; Tommasino, M.; Arbyn, M.; Franceschi, S. Eurogin Roadmap: Comparative epidemiology of HPV infection and associated cancers of the head and neck and cervix. Int. J. Cancer 2014, 134, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Nyitray, A.G.; Kreimer, A.R.; Pierce Campbell, C.M.; Goodman, M.T.; Sudenga, S.L.; Monsonego, J.; Franceschi, S. EUROGIN 2014 roadmap: Differences in human papillomavirus infection natural history, transmission and human papillomavirus-related cancer incidence by gender and anatomic site of infection. Int. J. Cancer 2014, 136, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Sepiashvili, L.; Bruce, J.P.; Huang, S.H.; O’Sullivan, B.; Liu, F.F.; Kislinger, T. Novel insights into head and neck cancer using next-generation “omic” technologies. Cancer Res. 2015, 75, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Lee, J.H.; Fulp, W.; Villa, L.L.; Lazcano, E.; Papenfuss, M.R.; Abrahamsen, M.; Salmeron, J.; Anic, G.M.; Rollison, D.E.; et al. Incidence and clearance of genital human papillomavirus infection in men (HIM): A cohort study. Lancet 2011, 377, 932–940. [Google Scholar] [CrossRef]
- Satterwhite, C.L.; Torrone, E.; Meites, E.; Dunne, E.F.; Mahajan, R.; Ocfemia, M.C.; Su, J.; Xu, F.; Weinstock, H. Sexually transmitted infections among US women and men: Prevalence and incidence estimates, 2008. Sex. Transm. Dis. 2013, 40, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, C.; Westra, W.H.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P.; Eriksen, J.G.; Hamilton-Dutoit, S.; Tramm, T.; Alsner, J.; Overgaard, J. Effect of HPV-associated p16INK4A expression on response to radiotherapy and survival in squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2009, 27, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan, B.; Huang, S.H.; Perez-Ordonez, B.; Massey, C.; Siu, L.L.; Weinreb, I.; Hope, A.; Kim, J.; Bayley, A.J.; Cummings, B.; et al. Outcomes of HPV-related oropharyngeal cancer patients treated by radiotherapy alone using altered fractionation. Radiother. Oncol. 2012, 103, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Gillison, M.L. Human papillomavirus in head and neck cancer: Its role in pathogenesis and clinical implications. Clin. Cancer Res. 2009, 15, 6758–6762. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Louvanto, K.; Rautava, J.; Willberg, J.; Wideman, L.; Syrjanen, K.; Grenman, S.; Syrjanen, S. Genotype-specific incidence and clearance of human papillomavirus in oral mucosa of women: A six-year follow-up study. PLoS ONE 2013, 8, e53413. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ordonez, B.; Beauchemin, M.; Jordan, R.C. Molecular biology of squamous cell carcinoma of the head and neck. J. Clin. Pathol. 2006, 59, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Hayes, D.N.; Grandis, J.R. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. 2015, 5, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Lleras, R.A.; Smith, R.V.; Adrien, L.R.; Schlecht, N.F.; Burk, R.D.; Harris, T.M.; Childs, G.; Prystowsky, M.B.; Belbin, T.J. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 5444–5455. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Parker, J.S.; Karaca, G.; Wu, J.; Funkhouser, W.K.; Moore, D.; Butterfoss, D.; Xiang, D.; Zanation, A.; Yin, X.; et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004, 5, 489–500. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Robinson, M.; Sloan, P.; Shaw, R. Refining the diagnosis of oropharyngeal squamous cell carcinoma using human papillomavirus testing. Oral Oncol. 2010, 46, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Abba, M.C.; Molinolo, A.A.; Vitale-Cross, L.; Wang, Z.; Zaida, M.; Delic, N.C.; Samuels, Y.; Lyons, J.G.; Gutkind, J.S. The head and neck cancer cell oncogenome: A platform for the development of precision molecular therapies. Oncotarget 2014, 5, 8906–8923. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Amelio, I.; Guerrero, U.T.; Tavassoli, M. Clinical update on cancer: Molecular oncology of head and neck cancer. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.; Baujat, B.; Holostenco, V.; Bourredjem, A.; Baey, C.; Bourhis, J.; Pignon, J.P. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): A comprehensive analysis by tumour site. Radiother. Oncol. 2011, 100, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Zhang, Q.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Sherman, E.J.; Weber, R.S.; Galvin, J.M.; Bonner, J.A.; Harris, J.; El-Naggar, A.K.; et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J. Clin. Oncol. 2014, 32, 2940–2950. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Zhang, Q.; Jordan, R.; Xiao, W.; Westra, W.H.; Trotti, A.; Spencer, S.; Harris, J.; Chung, C.H.; Ang, K.K. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol. 2012, 30, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Perrone, F.; Bossi, P.; Suardi, S.; Mariani, L.; Artusi, R.; Oggionni, M.; Rossini, C.; Cantu, G.; Squadrelli, M.; et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J. Clin. Oncol. 2006, 24, 5630–5636. [Google Scholar] [CrossRef] [PubMed]
- Posner, M.R.; Lorch, J.H.; Goloubeva, O.; Tan, M.; Schumaker, L.M.; Sarlis, N.J.; Haddad, R.I.; Cullen, K.J. Survival and human papillomavirus in oropharynx cancer in TAX 324: A subset analysis from an international phase III trial. Ann. Oncol. 2011, 22, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Rischin, D.; Young, R.J.; Fisher, R.; Fox, S.B.; Le, Q.T.; Peters, L.J.; Solomon, B.; Choi, J.; O’Sullivan, B.; Kenny, L.M.; et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J. Clin. Oncol. 2010, 28, 4142–4148. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Rampias, T.; Vermorken, J.B. The current and future impact of human papillomavirus on treatment of squamous cell carcinoma of the head and neck. Ann. Oncol. 2014, 25, 2101–2115. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Stohlmacher-Williams, J.; Davidenko, I.; Licitra, L.; Winquist, E.; Villanueva, C.; Foa, P.; Rottey, S.; Skladowski, K.; Tahara, M.; et al. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): An open-label phase 3 randomised trial. Lancet Oncol. 2013, 14, 697–710. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Psyrri, A.; Mesia, R.; Peyrade, F.; Beier, F.; de Blas, B.; Celik, I.; Licitra, L. Impact of tumor HPV status on outcome in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck receiving chemotherapy with or without cetuximab: Retrospective analysis of the phase III EXTREME trial. Ann. Oncol. 2014, 25, 801–807. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.; Huang, S.H.; Siu, L.L.; Waldron, J.; Zhao, H.; Perez-Ordonez, B.; Weinreb, I.; Kim, J.; Ringash, J.; Bayley, A.; et al. Deintensification candidate subgroups in human papillomavirus-related oropharyngeal cancer according to minimal risk of distant metastasis. J. Clin. Oncol. 2013, 31, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Vozenin, M.C.; Lord, H.K.; Hartl, D.; Deutsch, E. Unravelling the biology of human papillomavirus (HPV) related tumours to enhance their radiosensitivity. Cancer Treat. Rev. 2010, 36, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, D.W.; Spanos, W.C.; Vermeer, P.D.; Bruns, A.M.; Lee, K.M.; Lee, J.H. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int. J. Cancer 2013, 133, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Sikora, A.G.; Fu, S.; Kao, J. HPV-induced oropharyngeal cancer, immune response and response to therapy. Cancer Lett. 2010, 288, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Nowicki, P.; Lee, D.W.; Hoover, A.; Hostager, B.; Gupta, A.; Anderson, M.E.; Lee, J.H. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P.; Eriksen, J.G.; Hamilton-Dutoit, S.; Tramm, T.; Alsner, J.; Overgaard, J. HPV-associated p16-expression and response to hypoxic modification of radiotherapy in head and neck cancer. Radiother. Oncol. 2010, 94, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.S.; Narasimhan, B.; Cao, H.; Kwok, S.; Erickson, J.P.; Koong, A.; Pourmand, N.; Le, Q.T. The relationship between human papillomavirus status and other molecular prognostic markers in head and neck squamous cell carcinomas. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Toustrup, K.; Sorensen, B.S.; Nordsmark, M.; Busk, M.; Wiuf, C.; Alsner, J.; Overgaard, J. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. 2011, 71, 5923–5931. [Google Scholar] [CrossRef] [PubMed]
- Toustrup, K.; Sorensen, B.S.; Alsner, J.; Overgaard, J. Hypoxia gene expression signatures as prognostic and predictive markers in head and neck radiotherapy. Semin. Radiat. Oncol. 2012, 22, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Toustrup, K.; Sorensen, B.S.; Lassen, P.; Wiuf, C.; Alsner, J.; Overgaard, J. Gene expression classifier predicts for hypoxic modification of radiotherapy with nimorazole in squamous cell carcinomas of the head and neck. Radiother. Oncol. 2012, 102, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Hanns, E.; Job, S.; Coliat, P.; Wasylyk, C.; Ramolu, L.; Pencreach, E.; Suarez-Carmona, M.; Herfs, M.; Ledrappier, S.; Macabre, C.; et al. Human Papillomavirus-related tumours of the oropharynx display a lower tumour hypoxia signature. Oral Oncol. 2015, 51, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.; Delic, N.C.; Hong, A.; Zhang, M.; Rose, B.R.; Lyons, J.G. Radiosensitization of oropharyngeal squamous cell carcinoma cells by human papillomavirus 16 oncoprotein E6*I. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Arenz, A.; Ziemann, F.; Mayer, C.; Wittig, A.; Dreffke, K.; Preising, S.; Wagner, S.; Klussmann, J.P.; Engenhart-Cabillic, R.; Wittekindt, C. Increased radiosensitivity of HPV-positive head and neck cancer cell lines due to cell cycle dysregulation and induction of apoptosis. Strahlenther. Onkol. 2014, 190, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Gubanova, E.; Brown, B.; Ivanov, S.V.; Helleday, T.; Mills, G.B.; Yarbrough, W.G.; Issaeva, N. Downregulation of SMG-1 in HPV-positive head and neck squamous cell carcinoma due to promoter hypermethylation correlates with improved survival. Clin. Cancer Res. 2012, 18, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Tribius, S.; Grob, T.J.; Meyer, F.; Busch, C.J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Kalev, P.; van Limbergen, E.J.; Asbagh, L.A.; Vazquez, I.; Hauben, E.; Sablina, A.; Nuyts, S. p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res. 2014, 74, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S., Jr. p16 Immunohistochemistry as a standalone test for risk stratification in oropharyngeal squamous cell carcinoma. Head Neck Pathol. 2012, 6, S75–S82. [Google Scholar] [CrossRef] [PubMed]
- Lassen, P. The role of Human papillomavirus in head and neck cancer and the impact on radiotherapy outcome. Radiother. Oncol. 2010, 95, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.G.; Trivedi, T.I.; Tankshali, R.A.; Goswami, J.V.; Jetly, D.H.; Shukla, S.N.; Shah, P.M.; Verma, R.J. Prognostic significance of molecular markers in oral squamous cell carcinoma: A multivariate analysis. Head Neck 2009, 31, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Sedghizadeh, P.P.; Billington, W.D.; Paxton, D.; Ebeed, R.; Mahabady, S.; Clark, G.T.; Enciso, R. Is p16-positive oropharyngeal squamous cell carcinoma associated with favorable prognosis? A systematic review and meta-analysis. Oral Oncol. 2016, 54, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S., Jr.; Thorstad, W.L.; Chernock, R.D.; Haughey, B.H.; Yip, J.H.; Zhang, Q.; El-Mofty, S.K. p16 positive oropharyngeal squamous cell carcinoma: An entity with a favorable prognosis regardless of tumor HPV status. Am. J. Surg. Pathol. 2010, 34, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Gloghini, A.; Cortelazzi, B.; Bossi, P.; Licitra, L.; Pilotti, S. Isolating p16-positive/HPV-negative oropharyngeal cancer: An effort worth making. Am. J. Surg. Pathol. 2011, 35, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Richards, L. Genetics: Staging and prognostic value of p16 expression in oropharyngeal cancer. Nat. Rev. Clin. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Rietbergen, M.M.; Brakenhoff, R.H.; Bloemena, E.; Witte, B.I.; Snijders, P.J.; Heideman, D.A.; Boon, D.; Koljenovic, S.; Baatenburg-de Jong, R.J.; Leemans, C.R. Human papillomavirus detection and comorbidity: Critical issues in selection of patients with oropharyngeal cancer for treatment De-escalation trials. Ann. Oncol. 2013, 24, 2740–2745. [Google Scholar] [CrossRef] [PubMed]
- Nuyts, S.; Dirix, P.; Clement, P.M.; Poorten, V.V.; Delaere, P.; Schoenaers, J.; Hermans, R.; van den Bogaert, W. Impact of adding concomitant chemotherapy to hyperfractionated accelerated radiotherapy for advanced head-and-neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.P.; le Maître, A.; Maillard, E.; Bourhis, J. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 93 randomised trials and 17,346 patients. Radiother. Oncol. 2009, 92, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M. Radiobiological considerations in the design of clinical trials. Radiother. Oncol. 1994, 32, 1–11. [Google Scholar] [CrossRef]
- Masterson, L.; Moualed, D.; Liu, Z.W.; Howard, J.E.; Dwivedi, R.C.; Tysome, J.R.; Benson, R.; Sterling, J.C.; Sudhoff, H.; Jani, P.; et al. De-escalation treatment protocols for human papillomavirus-associated oropharyngeal squamous cell carcinoma: A systematic review and meta-analysis of current clinical trials. Eur. J. Cancer 2014, 50, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Harari, P.M. Is radiation dose reduction the right answer for HPV-positive head and neck cancer? Oral Oncol. 2014, 50, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Seiwert, T.Y.; Jimeno, A. Molecular pathways in head and neck cancer: EGFR, PI3K, and more. Am. Soc. Clin. Oncol. Educ. Book 2013, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Rosenthal, D.I.; Harari, P.M.; Giralt, J.; Bell, D.; Raben, D.; Liu, J.; Schulten, J.; Ang, K.K.; Bonner, J.A. Association of human papillomavirus and p16 status with outcomes in the IMCL-9815 phase III registration trial for patients with locoregionally advanced oropharyngeal squamous cell carcinoma of the head and neck treated with radiotherapy with or without cetuximab. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Abazeed, M.E.; Adams, D.J.; Hurov, K.E.; Tamayo, P.; Creighton, C.J.; Sonkin, D.; Giacomelli, A.O.; Du, C.; Fries, D.F.; Wong, K.K.; et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013, 73, 6289–6298. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, I. DNA repair inhibitors in cancer treatment. Clin. Transl. Oncol. 2006, 8, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Maxmen, A. Beyond PARP inhibitors: Agents in pipelines target DNA repair mechanisms. J. Natl. Cancer Inst. 2010, 102, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J.; Lakshman, M.; Chan, N.; Bristow, R.G. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin. Radiat. Oncol. 2010, 20, 274–281. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HPV Positive | HPV Negative | |
|---|---|---|
| Clinical, epidemiological characteristics | ||
| Incidence | Increasing | Decreasing |
| Age | Younger | Older |
| Socioeconomic status | Higher | Lower |
| Risk factors | Sexual behavior, marijuana exposure | Tobacco and alcohol exposure |
| Location of the tumor | Oropharynx (common in tonsil and BOT) | All head and neck sites (common in floor of mouth, lateral tongue and ventral tongue) |
| Prognosis | good | poor |
| Biological and histopathology characteristics | ||
| TP53 pathway | E6 mediated degradation | TP53 mutations |
| RB pathway | E7 mediated degradation | Inactivating mutations or other alterations in pathway |
| p16INK4a expression | Commonly overexpressed | Commonly decreased expression (inactivating mutations and hyper methylation) |
| Histology | Poorly differentiated or basaloid SCC | Modestly to well differentiated, keratinized SCC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dok, R.; Nuyts, S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers 2016, 8, 41. https://doi.org/10.3390/cancers8040041
Dok R, Nuyts S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers. 2016; 8(4):41. https://doi.org/10.3390/cancers8040041
Chicago/Turabian StyleDok, Rüveyda, and Sandra Nuyts. 2016. "HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies" Cancers 8, no. 4: 41. https://doi.org/10.3390/cancers8040041
APA StyleDok, R., & Nuyts, S. (2016). HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers, 8(4), 41. https://doi.org/10.3390/cancers8040041