
Mitochondrial Redox Signaling and Tumor Progression


{kind=link}
Abstract
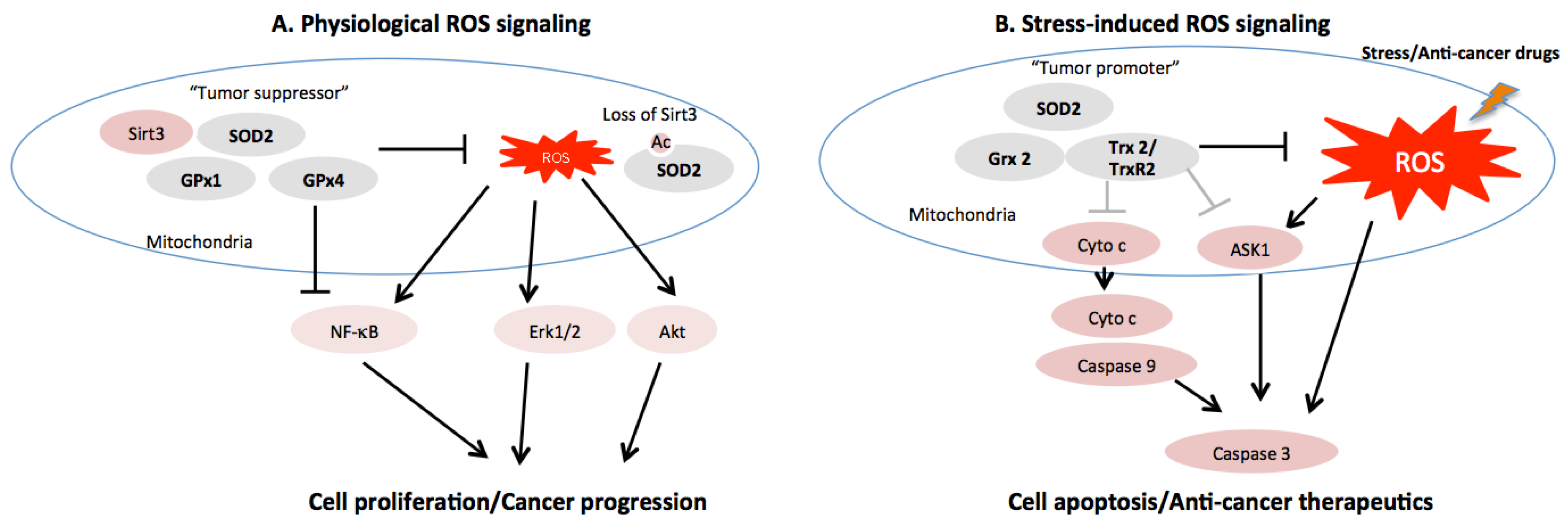
:1. Introduction
2. Manganese Superoxide Dismutase (SOD2)
3. Mitochondrial Glutaredoxin-2
4. Glutathione Peroxidase 1 (GPx-1) and Phospholipid Hydroperoxide GPx (GPx-4)
5. Thioredoxin 2 System
6. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AP-1 | activation protein-1 |
| ASK1 | apoptosis signal-regulating kinase 1 |
| EC | endothelial cell |
| ER | Endoplasmic reticulum |
| Grx2 | Glutaredoxin-2 |
| GPx | Glutathione peroxidase |
| NF-kB | Nuclear factor-kB |
| OXPHOS | Oxidative phosphorylation |
| Prx3 | Peroxidase-3 |
| ROS | Reactive oxidative species |
| SOD2 | superoxide dismutase |
| Trx2 | Thioredoixn-2 |
| TrxR2 | Trx2 reductase-2 |
References
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Teleman, A.A. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.S. The molecular biology of cancer. Mol. Aspects Med. 2000, 21, 167–223. [Google Scholar] [CrossRef]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr. Opin. Genet. Dev. 2010, 20, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peralta, S.; Moraes, C.T. Mitochondrial alterations during carcinogenesis: A review of metabolic transformation and targets for anticancer treatments. Adv. Cancer Res. 2013, 119, 127–160. [Google Scholar] [PubMed]
- Zhang, S.; Yang, C.; Yang, Z.; Zhang, D.; Ma, X.; Mills, G.; Liu, Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am. J. Cancer Res. 2015, 5, 928–944. [Google Scholar] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D'Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, A. Mitochondria in cancer energy metabolism: Culprits or bystanders? Toxicol. Res. 2015, 31, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Zamzami, N.; Susin, S.A.; Marchetti, P.; Hirsch, T.; Gómez-Monterrey, I.; Castedo, M.; Kroemer, G. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996, 183, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Han, V.; Han, J. New components of the necroptotic pathway. Protein Cell 2012, 3, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Giles, G.I. The redox regulation of thiol dependent signaling pathways in cancer. Curr. Pharm. Des. 2006, 12, 4427–4443. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Suzuki-Karasaki, Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic. Biol. Med. 2013, 61, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int. J. Mol. Med. 1999, 4, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.D.; Strieter, R.; Burdick, M.; Haghnegahdar, H.; Nanney, L.; Shattuck-Brandt, R.; Richmond, A. Enhanced tumor-forming capacity for immortalized melanocytes expressing melanoma growth stimulatory activity/growth-regulated cytokine beta and gamma proteins. Int. J. Cancer 1997, 73, 94–103. [Google Scholar] [CrossRef]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. MT1-MMP is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br. J. Cancer 2007, 97, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Antiangiogenic gene therapy. Proc. Natl. Acad. Sci. USA 1998, 95, 9064–9066. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Haudenschild, C.C.; Zetter, B.R. Long-term culture of capillary endothelial cells. Proc. Natl. Acad. Sci. USA 1979, 76, 5217–5721. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [PubMed]
- Gallogly, M.M.; Shelton, M.D.; Qanungo, S.; Pai, H.V.; Starke, D.W.; Hoppel, C.L.; Lesnefsky, E.J.; Mieyal, J.J. Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB targets Bcl-2 and Bcl-xL: Implications for cardiac aging. Antioxid. Redox Signal. 2010, 12, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.N.; Park, W.J.; Choi, Y.J.; Gurunathan, S.; Kim, J.H. Oxidative stress and ROS metabolism via down-regulation of sirtuin 3 expression in Cmah-null mice affect hearing loss. Aging (Albany NY) 2015, 7, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef] [PubMed]
- Knopp, E.A.; Arndt, T.L.; Eng, K.L.; Caldwell, M.; LeBoeuf, R.C.; Deeb, S.S.; O’Brien, K.D. Murine phospholipid hydroperoxide glutathione peroxidase: cDNA sequence, tissue expression, and mapping. Mamm. Genome 1999, 10, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Vizuete, A.; Damdimopoulos, A.E.; Spyrou, G. The mitochondrial thioredoxin system. Antioxid. Redox Signal. 2000, 2, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Bartz, R.R.; Suliman, H.B.; Piantadosi, C.A. Redox mechanisms of cardiomyocyte mitochondrial protection. Front. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- St. Clair, D.K.; Holland, J.C. Complementary DNA encoding human colon cancer manganese superoxide dismutase and the expression of its gene in human cells. Cancer Res. 1991, 51, 939–943. [Google Scholar] [PubMed]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W. Anticancer therapy by overexpression of superoxide dismutase. Antioxid. Redox Signal. 2001, 3, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Oberley, L.W.; Oberley, T.D.; Yan, T.; Domann, F.E.; St Clair, D.K. Inhibition of cell growth and sensitization to oxidative damage by overexpression of manganese superoxide dismutase in rat glioma cells. Cell Growth Differ. 1996, 7, 1175–1186. [Google Scholar] [PubMed]
- Li, J.J.; Oberley, L.W.; St Clair, D.K.; Ridnour, L.A.; Oberley, T.D. Phenotypic changes induced in human breast cancer cells by overexpression of manganese-containing superoxide dismutase. Oncogene 1995, 10, 1989–2000. [Google Scholar] [PubMed]
- Zhao, Y.; Chaiswing, L.; Oberley, T.D.; Batinic-Haberle, I.; St. Clair, W.; Epstein, C.J.; St. Clair, D. A mechanism-based antioxidant approach for the reduction of skin carcinogenesis. Cancer Res. 2005, 65, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Oberley, L.W.; Fan, M.; Colburn, N.H. Inhibition of AP-1 and NF-kappaB by manganese-containing superoxide dismutase in human breast cancer cells. FASEB J. 1998, 12, 1713–1723. [Google Scholar] [PubMed]
- Macmillan-Crow, L.A.; Cruthirds, D.L. Invited review: Manganese superoxide dismutase in disease. Free Radic. Res. 2001, 34, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Hemachandra, L.P.; Shin, D.H.; Dier, U.; Iuliano, J.N.; Engelberth, S.A.; Uusitalo, L.M.; Murphy, S.K.; Hempel, N. Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res. 2015, 75, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Li, J.J. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic. Biol. Med. 2008, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Murley, J.S.; Baker, K.L.; Miller, R.C.; Darga, T.E.; Weichselbaum, R.R.; Grdina, D.J. SOD2-mediated adaptive responses induced by low-dose ionizing radiation via TNF signaling and amifostine. Free Radic. Biol. Med. 2011, 51, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Delhalle, S.; Deregowski, V.; Benoit, V.; Merville, M.P.; Bours, V. NF-kappaB-dependent MnSOD expression protects adenocarcinoma cells from TNF-alpha-induced apoptosis. Oncogene 2002, 21, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.K.; Zhang, H.J.; Yan, T.; Oberley, L.W.; Aggarwal, B.B. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J. Biol. Chem. 1998, 273, 13245–13254. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalova, A.; Bikineyeva, A.; Ivanov, S.; Kirilyuk, I.A.; Grigor’ev, I.A.; Dikalov, S.I. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal. 2013, 19, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–627. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, W.; Cui, N.; Wu, Z.; Jiang, C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J. Biol. Chem. 2010, 285, 38641–38648. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. Js. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed]
- Diotte, N.M.; Xiong, Y.; Gao, J.; Chua, B.H.; Ho, Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta 2009, 1793, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; deKemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, L.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; deKemp, R.; da Silva, J.; et al. Vertebrate-specific glutaredoxin is essential for brain development. Proc. Natl. Acad. Sci. USA 2011, 108, 20532–20537. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Lönn, M.E.; Enoksson, M.; Fernandes, A.P.; Holmgren, A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc. Natl. Acad. Sci. USA 2004, 101, 13227–13232. [Google Scholar] [CrossRef] [PubMed]
- Enoksson, M.; Fernandes, A.P.; Prast, S.; Lillig, C.H.; Holmgren, A.; Orrenius, S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 2005, 327, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Finch, J.S.; Tome, M.E.; Kwei, K.A.; Bowden, G.T. Catalase reverses tumorigenicity in a malignant cell line by an epidermal growth factor receptor pathway. Free Radic. Biol. Med. 2006, 40, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Preston, T.J.; Muller, W.J.; Singh, G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J. Biol. Chem. 2001, 276, 9558–9564. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yan, T.; Yang, J.Q.; Oberley, T.D.; Oberley, L.W. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000, 60, 3927–3939. [Google Scholar] [PubMed]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Mu, X.; Jiang, C.; Yang, G.; Chen, H.; Xue, W. Single-nucleotide polymorphisms of GPX1 and MnSOD and susceptibility to bladder cancer: a systematic review and meta-analysis. Tumour. Biol. 2014, 35, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Canli, O.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Liang, H.; Gu, M.; Qi, W.; Walter, C.A.; Roberts, L.J.; Herman, B.; Richardson, A.; van Remmen, H. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 2004, 279, 55137–55146. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Lichti, U.F.; Carlson, B.A.; Cataisson, C.; Ryscavage, A.O.; Mikulec, C.; Conrad, M.; Fischer, S.M.; Hatfield, D.L.; Yuspa, S.H. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J. Investig. Dermatol. 2013, 133, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, J.; Zhang, Y.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in pancreatic cancer by overexpression of phospholipid hydroperoxide glutathione peroxidase. Hum. Gene Ther. 2006, 17, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; García-Cabezas, M.A.; Casado, E.; Belda-Iniesta, C.; de Castro, J.; Fresno, J.A.; Sereno, M.; Barriuso, J.; Espinosa, E.; Zamora, P.; et al. Phospholipid hydroperoxide glutathione peroxidase (PHGPx) expression is downregulated in poorly differentiated breast invasive ductal carcinoma. Free Radic. Res. 2007, 41, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Heirman, I.; Ginneberge, D.; Brigelius-Flohé, R.; Hendrickx, N.; Agostinis, P.; Brouckaert, P.; Rottiers, P.; Grooten, J. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic. Biol. Med. 2006, 40, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Brigelius-Flohe, R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005, 7, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Van Blarigan, E.L.; Ma, J.; Kenfield, S.A.; Stampfer, M.J.; Sesso, H.D.; Giovannucci, E.L.; Witte, J.S.; Erdman, J.W., Jr.; Chan, J.M.; Penney, K.L. Plasma antioxidants, genetic variation in SOD2, CAT, GPX1, GPX4, and prostate cancer survival. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, G.; Enmark, E.; Miranda-Vizuete, A.; Gustafsson, J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997, 272, 2936–2941. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Vizuete, A.; Damdimopoulos, A.E.; Spyrou, G. cDNA cloning, expression and chromosomal localization of the mouse mitochondrial thioredoxin reductase gene (1). Biochim. Biophys. Acta 1999, 1447, 113–118. [Google Scholar] [CrossRef]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pr. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Zhang, H.; Jones, D.P. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. 2006, 91, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Damdimopoulos, A.E.; Miranda-Vizuete, A.; Pelto-Huikko, M.; Gustafsson, J.A.; Spyrou, G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 2002, 277, 33249–33257. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Masutani, H.; Oka, S.; Tanaka, T.; Yamaguchi-Iwai, Y.; Nakamura, H.; Yodoi, J. Control of mitochondrial outer membrane permeabilization and Bcl-xL levels by thioredoxin 2 in DT40 cells. J. Biol. Chem. 2006, 281, 7384–7391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Damdimopoulos, A.E.; Spyrou, G.; Brune, B. Thioredoxin 1 and thioredoxin 2 have opposed regulatory functions on hypoxia-inducible factor-1alpha. J. Biol. Chem. 2007, 282, 7482–7490. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008, 320, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.I.; Lew, C.M.; Cortez, L.A.; Webb, C.R.; Rodriguez, M.; Liu, Y.; Qi, W.; Li, Y.; Chaudhuri, A.; van Remmen, H.; et al. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radic. Biol. Med. 2008, 44, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; He, Y.; Zhang, H.; Yu, L.; Wan, T.; Xu, Z.; Jones, D.; Chen, H.; Min, W. Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial ROS Generation and ASK1 Activity to Maintain Cardiac Function. Circulation 2015, 24, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Urig, S.; Becker, K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006, 16, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2016, 30, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Coronnello, M.; Mini, E.; Caciagli, B.; Cinellu, M.A.; Bindoli, A.; Gabbiani, C.; Messori, L. Mechanisms of cytotoxicity of selected organogold(III) compounds. J. Med. Chem. 2005, 48, 6761–6765. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Berggren, M.; Powis, G. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol. Cancer Res. 2003, 1, 682–689. [Google Scholar] [PubMed]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, H.J.; Huang, Q.; Lu, L.; Min, W. Novel action and mechanism of auranofin in inhibition of vascular endothelial growth factor receptor-3-dependent lymphangiogenesis. Anticancer Agents Med. Chem. 2014, 14, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, M.; Praggastis, A.; Li, Y.; He, Y.; Ghazvinian, R.; Cincotta, D.; Rice, K.; Min, W. Carbamoylating activity associated with the activation of the antitumor agent laromustine inhibits angiogenesis by inducing ASK1-dependent endothelial cell death. PLoS ONE 2014, 9, e103224. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, K.A.; Belousov, V.V. Genetically encoded fluorescent redox sensors. Biochim. Biophys. Acta 2014, 1840, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Ezerina, D.; Morgan, B.; Dick, T.P. Imaging dynamic redox processes with genetically encoded probes. J. Mol. Cell Cardiol. 2014, 73, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Heimer, N.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Redox proteomics: Methods for the identification and enrichment of redox-modified proteins and their applications. Proteomics 2016, 16, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhang, H.; Zhou, H.J.; Ji, W.; Min, W. Mitochondrial Redox Signaling and Tumor Progression. Cancers 2016, 8, 40. https://doi.org/10.3390/cancers8040040
Chen Y, Zhang H, Zhou HJ, Ji W, Min W. Mitochondrial Redox Signaling and Tumor Progression. Cancers. 2016; 8(4):40. https://doi.org/10.3390/cancers8040040
Chicago/Turabian StyleChen, Yuxin, Haiqing Zhang, Huanjiao Jenny Zhou, Weidong Ji, and Wang Min. 2016. "Mitochondrial Redox Signaling and Tumor Progression" Cancers 8, no. 4: 40. https://doi.org/10.3390/cancers8040040
APA StyleChen, Y., Zhang, H., Zhou, H. J., Ji, W., & Min, W. (2016). Mitochondrial Redox Signaling and Tumor Progression. Cancers, 8(4), 40. https://doi.org/10.3390/cancers8040040