Novel Molecular Challenges in Targeting Anaplastic Lymphoma Kinase in ALK-Expressing Human Cancers

Abstract
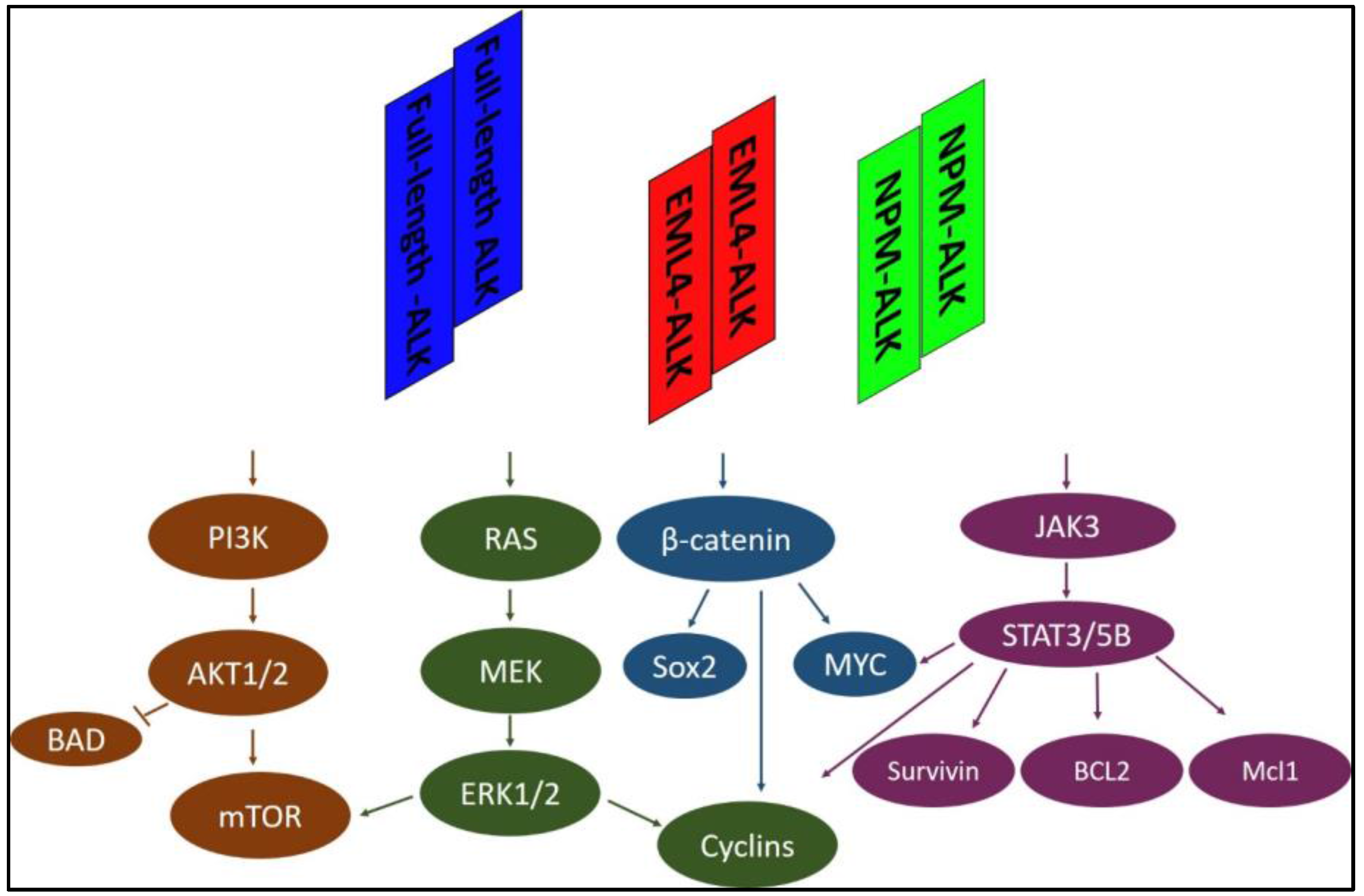
:1. Introduction
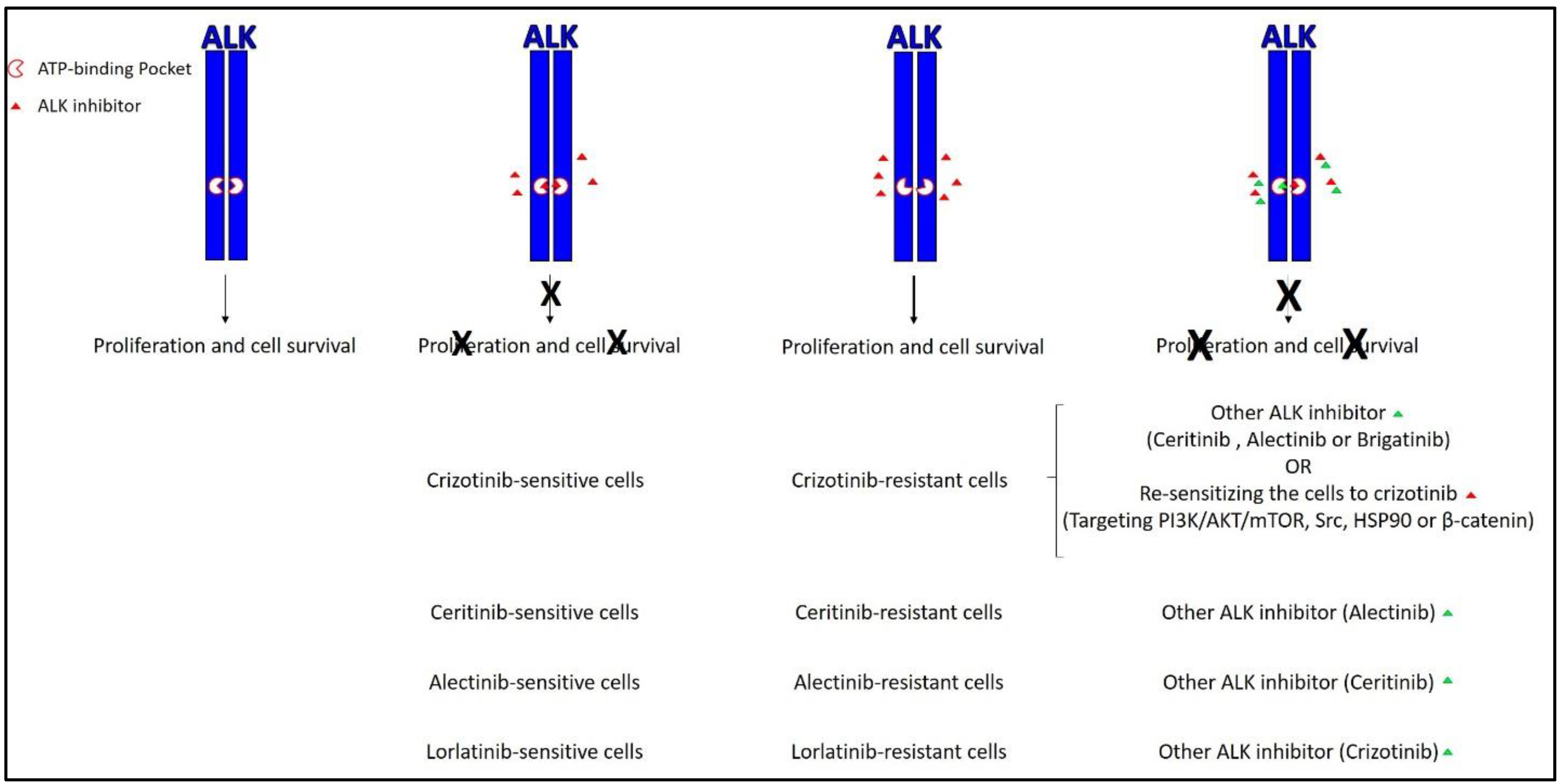
2. Reported Mechanisms of Resistance
3. Role of Intra-Tumoral Heterogeneity in Dictating Resistance to ALK Inhibitors in ALK-Expressing Cancers
4. Role of ALK-Interacting Proteins in Mediating TKIs Resistance
5. CETSA as a Tool That Can Be Used to Predict the Resistance to ALK Inhibitors
6. Potential Significance of Precursor mRNA in Mediating Resistance to ALK TKIs
7. Conclusions
Conflicts of Interest
References
- Vijayvergia, N.; Mehra, R. Clinical challenges in targeting anaplastic lymphoma kinase in advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2014, 74, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, J.; Semba, T.; Satoh, H.; Yamamoto, T.; Mori, S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 1994, 9, 1567–1574. [Google Scholar] [PubMed]
- Lai, R.; Ingham, R.J. The pathobiology of the oncogenic tyrosine kinase NPM-ALK: A brief update. Ther. Adv. Hematol. 2013, 4, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.M.; Lai, R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007, 110, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Grande, E.; Bolos, M.V.; Arriola, E. Targeting oncogenic ALK: A promising strategy for cancer treatment. Mol. Cancer Ther. 2011, 10, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., II; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Nat. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The Neuroblastoma-Associated F1174L ALK Mutation Causes Resistance to an ALK Kinase Inhibitor in ALK-Translocated Cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Azarova, A.M.; Gautam, G.; George, R.E. Emerging importance of ALK in neuroblastoma. Semin. Cancer Biol. 2011, 21, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Tiseo, M.; Di Maio, M.; Graziano, P.; Bria, E.; Rossi, G.; Novello, S. Tackling ALK in non-small cell lung cancer: The role of novel inhibitors. Transl. Lung Cancer Res. 2016, 5, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Engelman, J.A. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Ajimizu, H.; Kim, Y.H.; Mishima, M. Rapid response of brain metastases to alectinib in a patient with non-small-cell lung cancer resistant to crizotinib. Med. Oncol. 2015, 32, 477. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.F.; Azarova, A.M.; Bhatnagar, N.; Ross, K.N.; Drake, L.E.; Frumm, S.; Liu, Q.S.; Christie, A.L.; Sanda, T.; Chesler, L.; et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 8737–8749. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal. 2014, 7, ra102. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Alshareef, A.; Zhang, H.F.; Huang, Y.H.; Wu, C.; Zhang, J.D.; Wang, P.; El-Sehemy, A.; Fares, M.; Lai, R. The use of cellular thermal shift assay (CETSA) to study Crizotinib resistance in ALK-expressing human cancers. Sci. Rep. 2016, 6, 33710. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Milliken, J.C.; Azada, M.C.; Miller, V.A.; Ali, S.M.; Klempner, S.J. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer 2016, 91, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Greenbowe, J.; Khan, Z.U.; Azada, M.C.; Ross, J.S.; Stevens, P.J.; Ali, S.M.; Miller, V.A.; Gitlitz, B. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer 2015, 88, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-P.; Tsai, M.-F.; Chang, T.-H.; Tang, W.-C.; Chen, S.-Y.; Lai, H.-H.; Lin, T.-Y.; Yang, J.C.-H.; Yang, P.-C.; Shih, J.-Y.; et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013, 328, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Hashida, S.; Maki, Y.; Ichihara, E.; Asano, H.; et al. Acquired Resistance to EGFR Inhibitors Is Associated with a Manifestation of Stem cell-like Properties in Cancer Cells. Cancer Res. 2013, 73, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.J.; Palaiologou, D.; Panousopoulou, E.; Schultheis, B.; Yong, A.S.M.; Wong, A.; Pattacini, L.; Goldman, J.M.; Melo, J.V. Bcr-Abl Expression Levels Determine the Rate of Development of Resistance to Imatinib Mesylate in Chronic Myeloid Leukemia. Cancer Res. 2005, 65, 8912–8919. [Google Scholar] [CrossRef] [PubMed]
- Al-Achkar, W.; Wafa, A.; Moassass, F.; Klein, E.; Liehr, T. Multiple copies of BCR-ABL fusion gene on two isodicentric Philadelphia chromosomes in an imatinib mesylate-resistant chronic myeloid leukemia patient. Oncol. Lett. 2013, 5, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Zannettino, A.C.; Cambareri, A.C.; Quinn, S.R.; Manley, P.W.; Hughes, T.P. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): Reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 2006, 108, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhao, Y.; Smith, C.; Gasparetto, M.; Turhan, A.; Eaves, A.; Eaves, C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007, 21, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Traer, E.; MacKenzie, R.; Snead, J.; Agarwal, A.; Eiring, A.M.; O’Hare, T.; Druker, B.J.; Deininger, M.W. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia 2012, 26, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Wright, R.D.; McMillin, D.W.; Mitsiades, C.; Ray, A.; Barrett, R.; Adamia, S.; Stone, R.; Galinsky, I.; Kung, A.L.; et al. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol. Cancer Ther. 2008, 7, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Holtz, M.; Gupta, M.; Bhatia, R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood 2004, 103, 3167–3174. [Google Scholar] [CrossRef] [PubMed]
- Aceves-Luquero, C.I.; Agarwal, A.; Callejas-Valera, J.L.; Arias-Gonzalez, L.; Esparis-Ogando, A.; del Peso Ovalle, L.; Bellon-Echeverria, I.; de la Cruz-Morcillo, M.A.; Galan Moya, E.M.; Moreno Gimeno, I.; et al. ERK2, but not ERK1, mediates acquired and “de novo” resistance to imatinib mesylate: Implication for CML therapy. PLoS ONE 2009, 4, e6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar] [PubMed]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to SCLC after Treatment with the ALK Inhibitor Alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Lee, T.K.; Young, L.; Fernandez-Rocha, M.Y.; Pavlick, D.; Schrock, A.B.; Zhu, V.W.; Milliken, J.; Ali, S.M.; Gitlitz, B.J. Dual occurrence of ALK G1202R solvent front mutation and small cell lung cancer transformation as resistance mechanisms to second generation ALK inhibitors without prior exposure to crizotinib. Pitfall of solely relying on liquid re-biopsy? Lung Cancer 2017, 106, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALKF1174L-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Nicholes, K.; Bustos, D.; Lin, E.; Song, Q.; Stephan, J.P.; Kirkpatrick, D.S.; Settleman, J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014, 5, 7328–7341. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zang, J.; Qin, X.; Yan, D.; Cao, H.; Zhou, L.; Ni, J.; Yu, S.; Wu, J.; Feng, J.F. Epithelial-to-mesenchymal transition correlates with gefitinib resistance in NSCLC cells and the liver X receptor ligand GW3965 reverses gefitinib resistance through inhibition of vimentin. OncoTargets Ther. 2017, 10, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Soucheray, M.; Capelletti, M.; Pulido, I.; Kuang, Y.; Paweletz, C.P.; Becker, J.H.; Kikuchi, E.; Xu, C.; Patel, T.B.; Al-Shahrour, F.; et al. Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res. 2015, 75, 4372–4383. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.; Hsu, W.H.; Hsu, S.T.; Wang, Y.; Giaccone, G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol. Oncol. 2016, 10, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fu, L.-W. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit. Rev. Oncol. Hematol. 2015, 93, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Noh, K.H.; Lee, Y.-H.; Hong, S.-O.; Song, K.-H.; Lee, H.-J.; Kim, S.; Kim, T.M.; Jeon, J.-H.; Seo, J.H.; et al. Targeting stemness is an effective strategy to control EML4-ALK(+) non-small cell lung cancer cells. Oncotarget 2015, 6, 40255–40267. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897. [Google Scholar] [CrossRef] [PubMed]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALKF1174L Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Wu, C.; Alshareef, A.; Gupta, N.; Zhao, Q.; Xu, X.E.; Jiao, J.W.; Li, E.M.; Xu, L.Y.; Lai, R. The PI3K/AKT/c-MYC Axis Promotes the Acquisition of Cancer Stem-Like Features in Esophageal Squamous Cell Carcinoma. Stem Cells 2016, 34, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Gupta, N.; Wang, P.; Lewis, J.T.; Gopal, K.; Wu, F.; Ye, X.; Alshareef, A.; Abdulkarim, B.S.; Douglas, D.N.; et al. Triple negative breast cancers comprise a highly tumorigenic cell subpopulation detectable by its high responsiveness to a Sox2 regulatory region 2 (SRR2) reporter. Oncotarget 2015, 6, 10366–10373. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, J.; Wang, P.; Ye, X.; Jung, K.; Bone, K.M.; Pearson, J.D.; Ingham, R.J.; McMullen, T.P.; Ma, Y.; et al. Identification of two novel phenotypically distinct breast cancer cell subsets based on Sox2 transcription activity. Cell. Signal. 2012, 24, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Gelebart, P.; Hegazy, S.A.; Wang, P.; Bone, K.M.; Anand, M.; Sharon, D.; Hitt, M.; Pearson, J.D.; Ingham, R.J.; Ma, Y.; et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Furuhashi, M.; Nakane, R.; Nakazawa, S.; Goudarzi, H.; Hamada, J.-I.; Iizasa, H. Isolation and characterization of human breast cancer cells with SOX2 promoter activity. Biochem. Biophys. Res. Commun. 2013, 437, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.M.; Leis, O.; Pérez Ruiz, E.; Gumuzio Barrie, J.; Garcia-Garcia, F.; Aduriz, A.; Beloqui, I.; Hernandez-Garcia, S.; Lopez-Mato, M.P.; Dopazo, J.; et al. The Activation of the Sox2 RR2 Pluripotency Transcriptional Reporter in Human Breast Cancer Cell Lines is Dynamic and Labels Cells with Higher Tumorigenic Potential. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and Pharmacologic Inhibition of β-Catenin Targets Imatinib-Resistant Leukemia Stem Cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Nakagawa, K.; Nishio, K. Inhibition of β-Catenin enhances the anticancer effect of irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer with a T790M mutation. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hao, J. Development of anticancer agents targeting the Wnt/β-catenin signaling. Am. J. Cancer Res. 2015, 5, 2344–2360. [Google Scholar] [PubMed]
- Anand, M.; Lai, R.; Gelebart, P. β-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 2011, 96, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.J.; Martinez Molina, D.; Lundback, T. CETSA: A target engagement assay with potential to transform drug discovery. Future Med. Chem. 2015, 7, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Gokturk, C.; Sanjiv, K.; Stromberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, Y.; Maag, D.X.; Palma, J.P.; Patterson, M.J.; Ellis, P.A.; Surber, B.W.; Ready, D.B.; Soni, N.B.; Ladror, U.S.; et al. Iniparib Nonselectively Modifies Cysteine-Containing Proteins in Tumor Cells and Is Not a Bona Fide PARP Inhibitor. Clin. Cancer Res. 2012, 18, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. PARP inhibitors stumble in breast cancer. Nat. Biotech. 2011, 29, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Walker, D.; Bernardo, A.; Brodbeck, J.; Balestra, M.E.; Huang, Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Gladden, A.B.; Diehl, J.A. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003, 63, 7056–7061. [Google Scholar] [PubMed]
- Comstock, C.E.; Augello, M.A.; Benito, R.P.; Karch, J.; Tran, T.H.; Utama, F.E.; Tindall, E.A.; Wang, Y.; Burd, C.J.; Groh, E.M.; et al. Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. BioEssays News Rev. Mol. Cell. Dev. Boil. 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.-P.; et al. An Orally Available Small-Molecule Inhibitor of c-Met, PF-2341066, Exhibits Cytoreductive Antitumor Efficacy through Antiproliferative and Antiangiogenic Mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovly, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Oya, Y.; Tanaka, K.; Shimizu, J.; Horio, Y.; Kuroda, H.; Sakao, Y.; Hida, T.; Yatabe, Y. Differential Crizotinib Response Duration Among ALK Fusion Variants in ALK-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 3383–3389. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.G.; Seo, S.; Kim, S.W.; Jang, S.J.; Park, K.S.; Song, J.Y.; Lee, B.; Richards, M.W.; Bayliss, R.; Lee, D.H.; et al. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. The battle against ALK resistance: Successes and setbacks. Expert Opin. Invest. Drugs 2012, 21, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.-Y.; Yang, J.-J.; Zhong, W.-Z.; Chen, H.-J.; Yan, H.-H.; Han, J.-F.; Yang, L.-L.; Wu, Y.-L. Clinical efficacy of crizotinib in Chinese patients with ALK-positive non-small-cell lung cancer with brain metastases. J. Thorac. Dis. 2015, 7, 1181–1188. [Google Scholar] [PubMed]
- Zhou, Y.; Zhao, C.; Gery, S.; Braunstein, G.D.; Okamoto, R.; Alvarez, R.; Miles, S.A.; Doan, N.B.; Said, J.W.; Gu, J.; et al. Off-target effects of c-MET inhibitors on thyroid cancer cells. Mol. Cancer Ther. 2014, 13, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Shaw, A.T. ALK inhibitors in non-small cell lung cancer: Crizotinib and beyond. Clin. Adv. Hematol. Oncol. 2014, 12, 429–439. [Google Scholar]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med. Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet. Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Cortes, J. Flying under the radar: The new wave of BCR-ABL inhibitors. Nat. Rev. Drug Discov. 2007, 6, 834–848. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ALK Inhibitor | Other Names | FDA Approval (Month/Year) | Resistance Occurred [Reference] | Ways to Overcome Resistance [Reference] | |
|---|---|---|---|---|---|
| Other ALK Inhibitors | Re-Sensitizing the Inhibitor | ||||
| Crizotinib | PF-2341066 Xalkori® | Yes (08/2011) | Yes [15,23] | 1. Ceritinib [24,25] 2. Alectinib [26] 3. Brigatinib [27] | 1. Targeting PI3K/AKT/mTOR pathway [28,29] 2. Targeting Src [30] 3. Targeting HSP90 [31] 4. Targeting β-catenin [32] |
| Ceritinib | LDK-378 Zycadia® | Yes (04/2014) | Yes [33,34] | Alectinib [33,34] | Not performed |
| Alectinib | CH5424802 RO5424802 Alecensa® | Yes (12/2015) | Yes [35,36] | Ceritinib [35,37] | Not performed |
| Brigatinib | AP26113 Alunbrig™ | Yes (04/2017) | Yes [38] | - | Not performed |
| Lorlatinib | PF-06463922 | No | Yes [39] | Crizotinib [39] | Not performed |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshareef, A. Novel Molecular Challenges in Targeting Anaplastic Lymphoma Kinase in ALK-Expressing Human Cancers. Cancers 2017, 9, 148. https://doi.org/10.3390/cancers9110148
Alshareef A. Novel Molecular Challenges in Targeting Anaplastic Lymphoma Kinase in ALK-Expressing Human Cancers. Cancers. 2017; 9(11):148. https://doi.org/10.3390/cancers9110148
Chicago/Turabian StyleAlshareef, Abdulraheem. 2017. "Novel Molecular Challenges in Targeting Anaplastic Lymphoma Kinase in ALK-Expressing Human Cancers" Cancers 9, no. 11: 148. https://doi.org/10.3390/cancers9110148
APA StyleAlshareef, A. (2017). Novel Molecular Challenges in Targeting Anaplastic Lymphoma Kinase in ALK-Expressing Human Cancers. Cancers, 9(11), 148. https://doi.org/10.3390/cancers9110148