Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
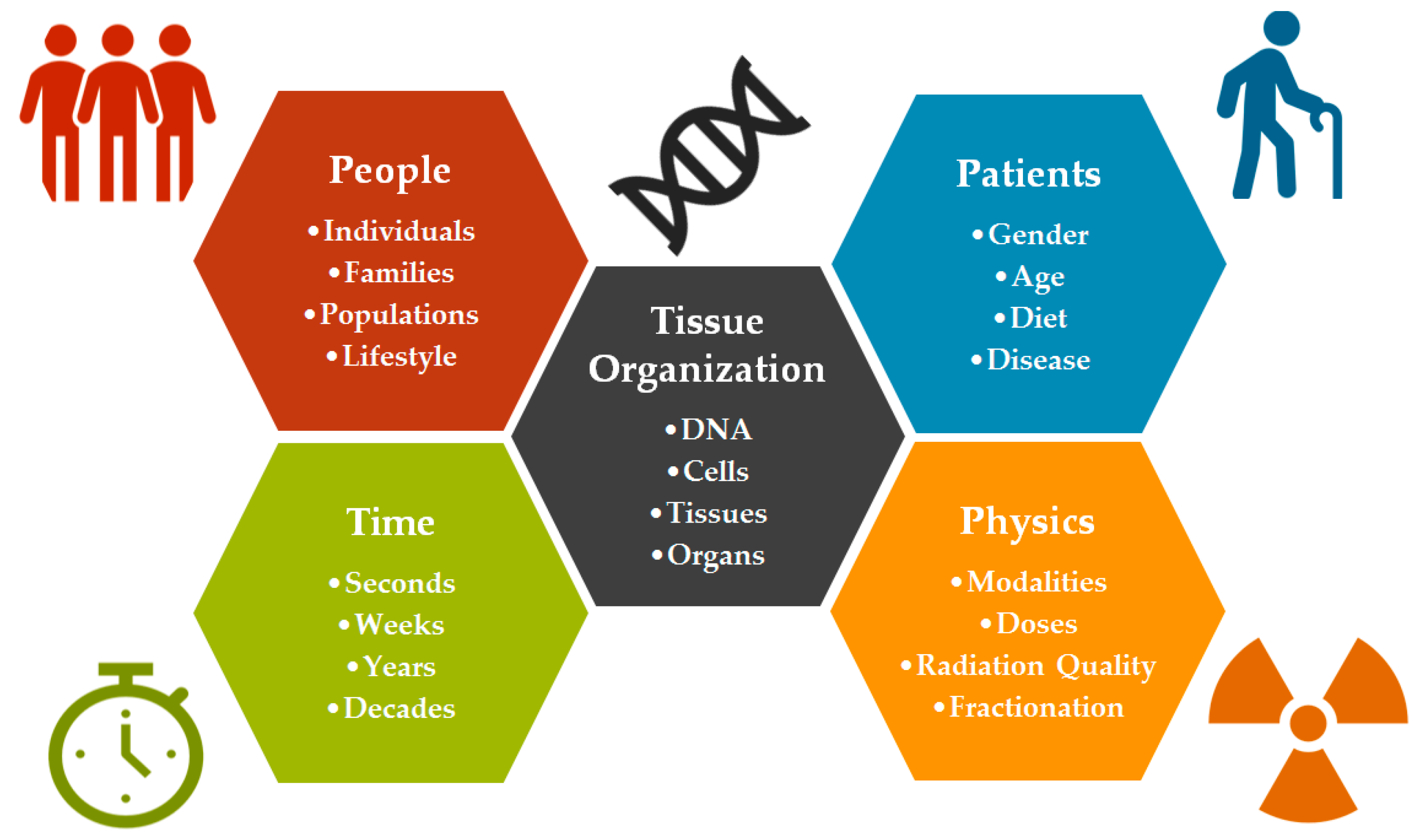
:1. Introduction
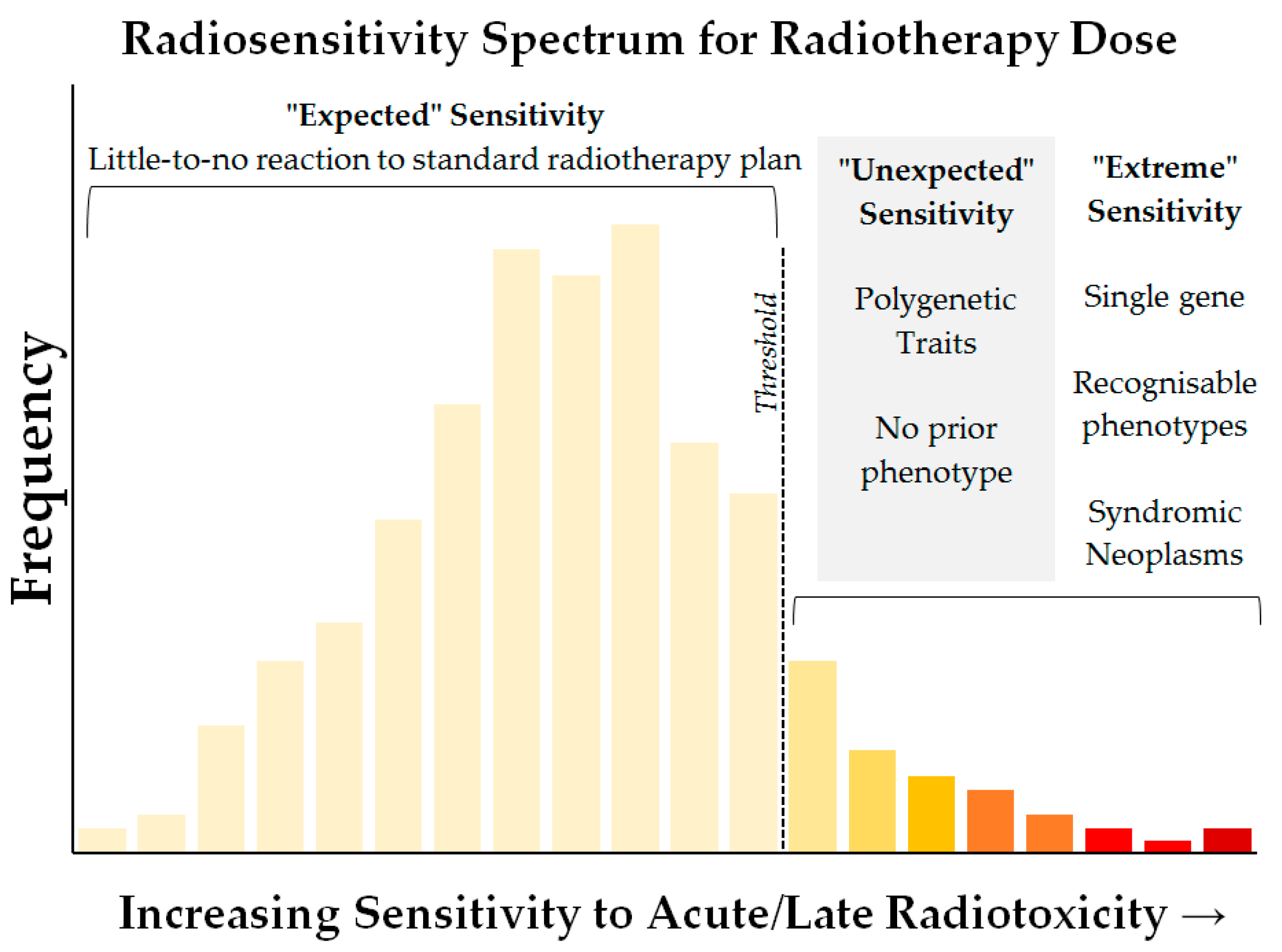
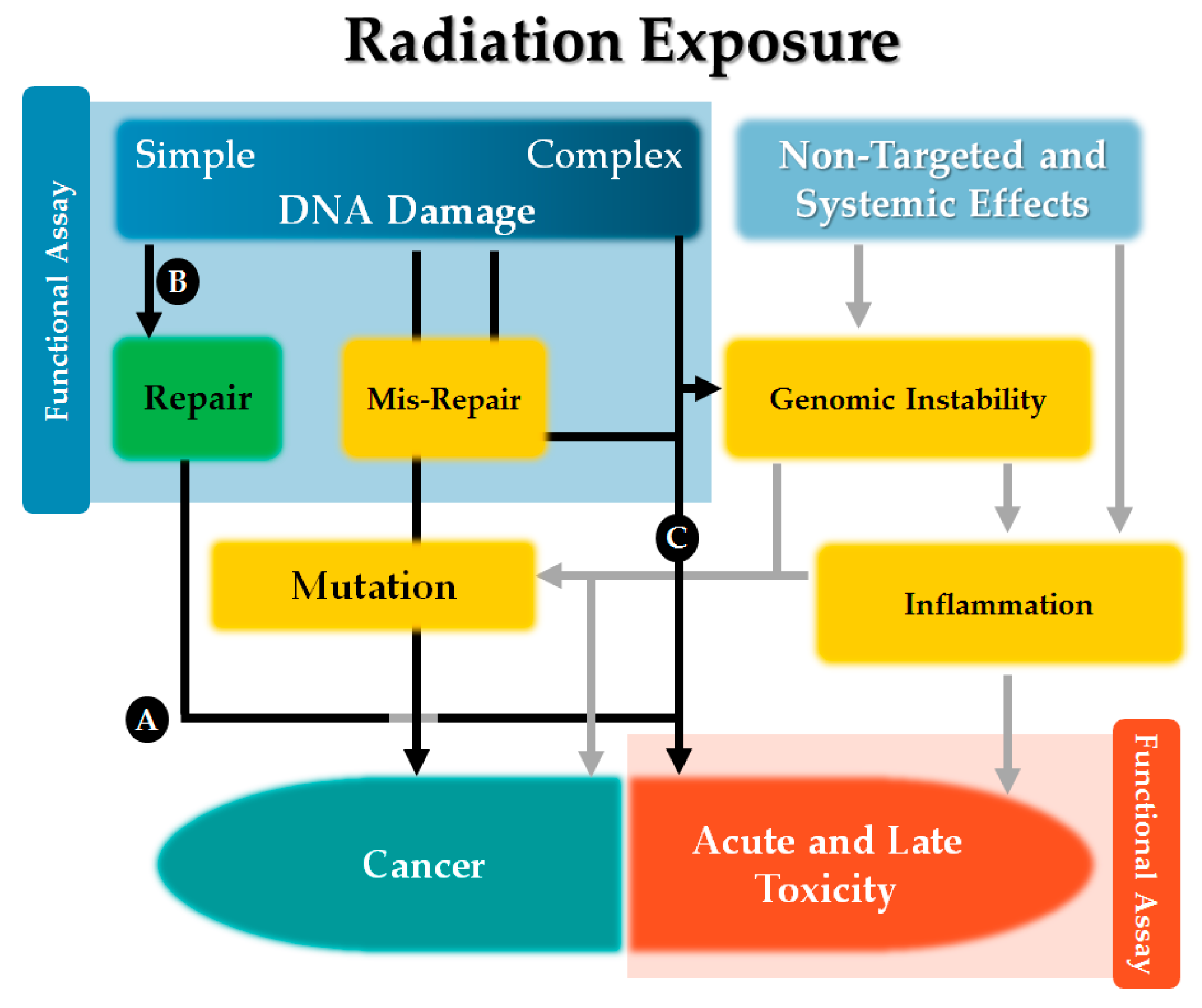
2. Observed Susceptibility to Radiation-Induced Toxicity
3. Observed Susceptibility to Radiation-Induced Cancer
4. Genetic Variation in Sensitivity to Radiation-Induced Acute and Late Toxicity
5. Genetic Variation in Sensitivity to Radiation-Induced Cancer
6. Selection of a Functional Assay
7. Opportunities for Risk Mitigation in Sensitive Patients
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kumar, S. Second malignant neoplasms following radiotherapy. Int. J. Environ. Res. Public Health 2012, 9, 4744–4759. [Google Scholar] [CrossRef] [PubMed]
- Berrington de Gonzalez, A.; Curtis, R.E.; Kry, S.F.; Gilbert, E.; Lamart, S.; Berg, C.D.; Stovall, M.; Ron, E. Proportion of second cancers attributable to radiotherapy treatment in adults: A cohort study in the US SEER cancer registries. Lancet Oncol. 2011, 12, 353–360. [Google Scholar] [CrossRef]
- Yock, T.I.; Caruso, P.A. Risk of second cancers after photon and proton radiotherapy: A review of the data. Health Phys. 2012, 103, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.K.; LaCasce, A.; Travis, L.B. Risk of second malignant neoplasm among patients with lymphoma reply. J. Clin. Oncol. 2011, 29, 3834–3835. [Google Scholar] [CrossRef]
- Marcu, L.G. Photons—Radiobiological issues related to the risk of second malignancies. Phys. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Herskind, C.; Talbot, C.J.; Kerns, S.L.; Veldwijk, M.R.; Rosenstein, B.S.; West, C.M.L. Radiogenomics: A systems biology approach to understanding genetic risk factors for radiotherapy toxicity? Cancer Lett. 2016, 382, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Barnett, G.C.; West, C.M.; Coles, C.E.; Pharoah, P.D.; Talbot, C.J.; Elliott, R.M.; Tanteles, G.A.; Symonds, R.P.; Wilkinson, J.S.; Dunning, A.M.; et al. Standardized total average toxicity score: A scale- and grade-independent measure of late radiotherapy toxicity to facilitate pooling of data from different studies. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Bouffler, S.D. Evidence for variation in human radiosensitivity and its potential impact on radiological protection. Ann. ICRP 2016, 45 (Suppl. 1), 280–289. [Google Scholar] [CrossRef] [PubMed]
- Stovall, M.; Smith, S.A.; Langholz, B.M.; Boice, J.D., Jr.; Shore, R.E.; Andersson, M.; Buchholz, T.A.; Capanu, M.; Bernstein, L.; Lynch, C.F.; et al. Dose to the contralateral breast from radiotherapy and risk of second primary breast cancer in the WECARE study. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Abo-Madyan, Y.; Aziz, M.H.; Aly, M.M.; Schneider, F.; Sperk, E.; Clausen, S.; Giordano, F.A.; Herskind, C.; Steil, V.; Wenz, F.; et al. Second cancer risk after 3D-CRT, IMRT and VMAT for breast cancer. Radiother. Oncol. 2014, 110, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Mansur, D.B.; Klein, E.E.; Maserang, B.P. Measured peripheral dose in pediatric radiation therapy: A comparison of intensity-modulated and conformal techniques. Radiother. Oncol. 2007, 82, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.G.; Bednarz, B.; Paganetti, H. A review of dosimetry studies on external-beam radiation treatment with respect to second cancer induction. Phys. Med. Biol. 2008, 53, R193–R241. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J. What we know and what we don’t know about cancer risks associated with radiation doses from radiological imaging. Br. J. Radiol. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Sachs, R.K.; Brenner, D.J. Solid tumor risks after high doses of ionizing radiation. Proc. Natl. Acad. Sci. USA. 2005, 102, 13040–13045. [Google Scholar] [CrossRef] [PubMed]
- Preston, D.L.; Ron, E.; Tokuoka, S.; Funamoto, S.; Nishi, N.; Soda, M.; Mabuchi, K.; Kodama, K. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat. Res. 2007, 181, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Valentin, J. Low-dose extrapolation of radiation-related cancer risk. Ann. ICRP 2005, 35, 1–140. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Health Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII, Phase II; National Research Council: Washington, DC, USA, 2006. [Google Scholar]
- Portess, D.I.; Bauer, G.; Hill, M.A.; O’Neill, P. Low-dose irradiation of nontransformed cells stimulates the selective removal of precancerous cells via intercellular induction of apoptosis. Cancer Res. 2007, 67, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.; Cordes, N. Radiation therapy and the microenvironment. Int. J. Radiat. Biol. 2009, 83, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Sachs, R.K.; Shuryak, I.; Brenner, D.; Fakir, H.; Hlatky, L.; Hahnfeldt, P. Second cancers after fractionated radiotherapy: Stochastic population dynamics effects. J. Theor. Biol. 2007, 249, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Sykes, P.J. Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiat. Res. 2011, 181, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Shuryak, I.; Sachs, R.K.; Brenner, D.J. Cancer risks after radiation exposure in middle age. J. Natl. Cancer Inst. 2010, 102, 1628–1636. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Curtis, R.E.; Hall, E.J.; Ron, E. Second malignancies in prostate carcinoma patients after radiotherapy compared with surgery. Cancer 2000, 88, 398–406. [Google Scholar] [CrossRef]
- Travis, L.B.; Demark Wahnefried, W.; Allan, J.M.; Wood, M.E.; Ng, A.K. Aetiology, genetics and prevention of secondary neoplasms in adult cancer survivors. Nat. Rev. Clin. Oncol. 2013, 10, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.L.; Armstrong, G.T.; Boice, J.D.; Chow, E.J.; Davies, S.M.; Donaldson, S.S.; Green, D.M.; Hammond, S.; Meadows, A.T.; Mertens, A.C.; et al. The childhood cancer survivor study: A national cancer institute–supported resource for outcome and intervention research. J. Clin. Oncol. 2009, 27, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Neglia, J.P.; Friedman, D.L.; Yasui, Y.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L. Second malignant neoplasms in five-year survivors of childhood cancer: Childhood cancer survivor study. J. Natl. Cancer Inst. 2001, 93, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Liu, Y.; Neglia, J.P.; Friedman, D.L.; Bhatia, S.; Meadows, A.T.; Diller, L.R.; Mertens, A.C.; Whitton, J.; Robison, L.L. A methodological issue in the analysis of second-primary cancer incidence in long-term survivors of childhood cancers. Am. J. Epidemiol. 2003, 158, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Robison, L.L.; Oberlin, O.; Greenberg, M.; Bunin, G.; Fossati-Bellani, F.; Meadows, A.T. Breast cancer and other second neoplasms after childhood hodgkin’s disease. N. Engl. J. Med. 1996, 334, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Neglia, J.P.; Robison, L.L.; Stovall, M.; Liu, Y.; Packer, R.J.; Hammond, S.; Yasui, Y.; Kasper, C.E.; Mertens, A.C.; Donaldson, S.S.; et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: A report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2006, 98, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.K.; Kenney, L.B.; Gilbert, E.S.; Travis, L.B. Secondary malignancies across the age spectrum. Semin. Radiat. Oncol. 2010, 20, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.E.; Vogel, V.; Ng, A.; Foxhall, L.; Goodwin, P.; Travis, L.B. Second malignant neoplasms: Assessment and strategies for risk reduction. J. Clin. Oncol. 2012, 30, 3734–3745. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, M. Can we reduce the incidence of second primary malignancies occurring after radiotherapy? A critical review. Radiother. Oncol. 2009, 91, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Inskip, P.D.; Sigurdson, A.J.; Veiga, L.; Bhatti, P.; Ronckers, C.; Rajaraman, P.; Boukheris, H.; Stovall, M.; Smith, S.; Hammond, S.; et al. Radiation-related new primary solid cancers in the childhood cancer survivor study: Comparative radiation dose response and modification of treatment effects. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.; Henry, A.; Hoskin, P.; Siebert, F.A.; Venselaar, J.; Estro, P.G.O.G. Second primary cancers after radiation for prostate cancer: A systematic review of the clinical data and impact of treatment technique. Radiother. Oncol. 2014, 110, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, C.; Blyth, B.J.; Morioka, T.; Kaminishi, M.; Shinagawa, M.; Shimada, Y.; Kakinuma, S. Sensitive detection of radiation-induced medulloblastomas after acute or protracted γ-ray exposures in PTCH1 heterozygous mice using a radiation-specific molecular signature. Radiat. Res. 2016, 186. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Kakinuma, S.; Sunaoshi, M.; Amasaki, Y.; Hirano-Sakairi, S.; Ogawa, K.; Shirakami, A.; Shang, Y.; Tsuruoka, C.; Nishimura, M.; et al. Genetic analysis of T cell lymphomas in carbon ion-irradiated mice reveals frequent interstitial chromosome deletions: Implications for second cancer induction in normal tissues during carbon ion radiotherapy. PLoS ONE 2015, 10, e0130666. [Google Scholar] [CrossRef] [PubMed]
- Rosemann, M.; Kuosaite, V.; Nathrath, M.; Atkinson, M.J. The genetics of radiation-induced osteosarcoma. Radiat. Prot. Dosim. 2002, 99, 257–259. [Google Scholar] [CrossRef]
- Stewart, F.A.; Dorr, W. Milestones in normal tissue radiation biology over the past 50 years: From clonogenic cell survival to cytokine networks and back to stem cell recovery. Int. J. Radiat. Biol. 2009, 85, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Braselmann, H.; Geinitz, H.; Jaehnert, I.; Baumgartner, A.; Thamm, R.; Figel, M.; Molls, M.; Zitzelsberger, H. Chromosomal radiosensitivity and acute radiation side effects after radiotherapy in tumour patients—A follow-up study. Radiat. Oncol. 2011, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.; Lavin, M.F. Cellular radiosensitivity: How much better do we understand it? Int. J. Radiat. Biol. 2009, 85, 1061–1081. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, A.; Thierens, H.; Vandenbulcke, K.; De Ridder, L.; Vral, A. The use of EBV-transformed cell lines of breast cancer patients to measure chromosomal radiosensitivity. Mutagenesis 2004, 19, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Greve, B.; Bolling, T.; Amler, S.; Rossler, U.; Gomolka, M.; Mayer, C.; Popanda, O.; Dreffke, K.; Rickinger, A.; Fritz, E.; et al. Evaluation of different biomarkers to predict individual radiosensitivity in an inter-laboratory comparison—Lessons for future studies. PLoS ONE 2012, 7, e47185. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Popanda, O.; Greve, B.; Fritz, E.; Illig, T.; Eckardt-Schupp, F.; Gomolka, M.; Benner, A.; Schmezer, P. A radiation-induced gene expression signature as a tool to predict acute radiotherapy-induced adverse side effects. Cancer Lett. 2011, 302, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Geara, F.B.; Peters, L.J.; Ang, K.K.; Wike, J.L.; Brock, W.A. Radiosensitivity measurement of keratinocytes and fibroblasts from radiotherapy patients. Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 287–293. [Google Scholar] [CrossRef]
- Geara, F.B.; Peters, L.J.; Ang, K.K.; Wike, J.L.; Sivon, S.S.; Guttenberger, R.; Callender, D.L.; Malaise, E.P.; Brock, W.A. Intrinsic radiosensitivity of normal human fibroblasts and lymphocytes after high- and low-dose-rate irradiation. Cancer Res. 1992, 52, 6348–6352. [Google Scholar] [PubMed]
- Finnon, P.; Robertson, N.; Dziwura, S.; Raffy, C.; Zhang, W.; Ainsbury, L.; Kaprio, J.; Badie, C.; Bouffler, S. Evidence for significant heritability of apoptotic and cell cycle responses to ionising radiation. Hum. Genet. 2008, 123, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Surowy, H.; Rinckleb, A.; Luedeke, M.; Stuber, M.; Wecker, A.; Varga, D.; Maier, C.; Hoegel, J.; Vogel, W. Heritability of baseline and induced micronucleus frequencies. Mutagenesis 2011, 26, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Curwen, G.B.; Cadwell, K.K.; Winther, J.F.; Tawn, J.E.; Rees, G.S.; Olsen, J.H.; Rechnitzer, C.; Schroeder, H.; Guldberg, P.; Cordell, H.J.; et al. The heritability of G2 chromosomal radiosensitivity and its association with cancer in Danish cancer survivors and their offspring. Int. J. Radiat. Biol. 2010, 86, 986–995. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.; Barnett, G.C. Genetics and genomics of radiotherapy toxicity: Towards prediction. Genome Med. 2011, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Bedford, J.S. Studies on chromosome aberration induction: What can they tell us about DNA repair? DNA Repair 2006, 5, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Keller, U.; Sprung, C.N.; Weise, A.; Grabenbauer, G.G.; Sauer, R.; Distel, L. Breakpoint locations within chromosomes 1, 2, and 4 of patients with increased radiosensitivity. Cancer Genet. Cytogenet. 2006, 168, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beaton, L.A.; Marro, L.; Samiee, S.; Malone, S.; Grimes, S.; Malone, K.; Wilkins, R.C. Investigating chromosome damage using fluorescent in situ hybridization to identify biomarkers of radiosensitivity in prostate cancer patients. Int. J. Radiat. Biol. 2013, 89, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Beaton, L.A.; Ferrarotto, C.; Marro, L.; Samiee, S.; Malone, S.; Grimes, S.; Malone, K.; Wilkins, R.C. Chromosome damage and cell proliferation rates in in vitro irradiated whole blood as markers of late radiation toxicity after radiation therapy to the prostate. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Distel, L.V.; Neubauer, S.; Keller, U.; Sprung, C.N.; Sauer, R.; Grabenbauer, G.G. Individual differences in chromosomal aberrations after in vitro irradiation of cells from healthy individuals, cancer and cancer susceptibility syndrome patients. Radiother. Oncol. 2006, 81, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Eastham, A.M.; Marples, B.; Kiltie, A.E.; Orton, C.J.; West, C.M. Fibroblast radiosensitivity measured using the comet DNA-damage assay correlates with clonogenic survival parameters. Br. J. Cancer 1999, 79, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Kunogi, H.; Sakanishi, T.; Sueyoshi, N.; Sasai, K. Prediction of radiosensitivity using phosphorylation of histone H2AX and apoptosis in human tumor cell lines. Int. J. Radiat. Biol. 2014, 90, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Ivashkevich, A.; Choo, S.; Woodbine, L.; Jeggo, P.A.; Martin, R.F.; Lobachevsky, P. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: Cell cycle effect and implications for prediction of radiosensitivity. DNA Repair 2013, 12, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Lobachevsky, P.; Woodbine, L.; Hsiao, K.C.; Choo, S.; Fraser, C.; Gray, P.; Smith, J.; Best, N.; Munforte, L.; Korneeva, E.; et al. Evaluation of severe combined immunodeficiency and combined immunodeficiency pediatric patients on the basis of cellular radiosensitivity. J. Mol. Diagn. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Severin, D.; Murray, D. Relationship between DNA double-strand break rejoining and cell survival after exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: Implications for evaluation of clinical radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Joubert, A.; Zimmerman, K.M.; Bencokova, Z.; Gastaldo, J.; Chavaudra, N.; Favaudon, V.; Arlett, C.F.; Foray, N. DNA double-strand break repair defects in syndromes associated with acute radiation response: At least two different assays to predict intrinsic radiosensitivity? Int. J. Radiat. Biol. 2009, 84, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Kmochova, A.; Tichy, A.; Zarybnicka, L.; Sinkorova, Z.; Vavrova, J.; Rehacek, V.; Durisova, K.; Kubelkova, K.; Pejchal, J.; Kuca, K. Modulation of ionizing radiation-induced effects by NU7441, KU55933 and VE821 in peripheral blood lymphocytes. J. Appl. Biomed. 2016, 14, 19–24. [Google Scholar] [CrossRef]
- Mumbrekar, K.; Goutham, H.; Vadhiraja, B.; Sadashiva, S. Polymorphisms in double strand break repair related genes influence radiosensitivity phenotype in lymphocytes from healthy individuals. DNA Repair 2016, 40, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.; Bayer, J.; Dechamps, N.; Goldin, L.; Thomas, G. Heritability of susceptibility to ionizing radiation-induced apoptosis of human lymphocyte subpopulations. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Baijer, J.; Déchamps, N.; Perdry, H.; Morales, P.; Kerns, S.; Vasilescu, A.; Baulande, S.; Azria, D.; Roméo, P.; Schmitz, A. TNFSF10/TRAIL regulates human T4 effector memory lymphocyte radiosensitivity and predicts radiation-induced acute and subacute dermatitis. Oncotarget 2016, 7, 21416–21427. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Qin, Y.; Fridley, B.L.; Hou, J.; Kalari, K.R.; Zhu, M.; Wu, T.-Y.; Jenkins, G.D.; Batzler, A.; Wang, L. Radiation pharmacogenomics: A genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res. 2010, 20, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.L.; Ostrer, H.; Rosenstein, B.S. Radiogenomics: Using genetics to identify cancer patients at risk for development of adverse effects following radiotherapy. Cancer Discov. 2014, 4, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Fachal, L.; Gomez-Caamano, A.; Barnett, G.C.; Peleteiro, P.; Carballo, A.M.; Calvo-Crespo, P.; Kerns, S.L.; Sanchez-Garcia, M.; Lobato-Busto, R.; Dorling, L.; et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat. Genet. 2014, 46, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Barnett, G.C.; Thompson, D.; Fachal, L.; Kerns, S.; Talbot, C.; Elliott, R.M.; Dorling, L.; Coles, C.E.; Dearnaley, D.P.; Rosenstein, B.S.; et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother. Oncol. 2014, 111, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Rattay, T.; Talbot, C.J. Finding the genetic determinants of adverse reactions to radiotherapy. Clin. Oncol. 2014, 26, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.S.; Grummels, A.; Hart, A.A.; Smolders, I.J.; Borger, J.; Bartelink, H.; Begg, A.C. Low predictive value of intrinsic fibroblast radiosensitivity for fibrosis development following radiotherapy for breast cancer. Int. J. Radiat. Biol. 1998, 73, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Badie, C.; Dziwura, S.; Raffy, C.; Tsigani, T.; Alsbeih, G.; Moody, J.; Finnon, P.; Levine, E.; Scott, D.; Bouffler, S. Aberrant CDKN1A transcriptional response associates with abnormal sensitivity to radiation treatment. Br. J. Cancer 2008, 98, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, C.N.; Alsner, J.; Overgaard, M.; Sørensen, F.B.; Overgaard, J. Risk of radiation-induced subcutaneous fibrosis in relation to single nucleotide polymorphisms in TGFB1, SOD2, XRCC1, XRCC3, APEX and ATMndash: A study based on DNA from formalin fixed paraffin embedded tissue samples. Int. J. Radiat. Biol. 2009, 82, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Djuzenova, C.S.; Elsner, I.; Katzer, A.; Worschech, E.; Distel, L.V.; Flentje, M.; Polat, B. Radiosensitivity in breast cancer assessed by the histone γ-H2AX and 53BP1 foci. Radiat. Oncol. 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Finnon, P.; Kabacik, S.; MacKay, A.; Raffy, C.; A’Hern, R.; Owen, R.; Badie, C.; Yarnold, J.; Bouffler, S. Correlation of in vitro lymphocyte radiosensitivity and gene expression with late normal tissue reactions following curative radiotherapy for breast cancer. Radiother. Oncol. 2012, 105, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Hernández, L.; Carmona-Vigo, R.; Pinar, B.; Bordón, E.; Lloret, M.; Núñez, M.; Rodríguez-Gallego, C.; Lara, P.C. Combined low initial DNA damage and high radiation-induced apoptosis confers clinical resistance to long-term toxicity in breast cancer patients treated with high-dose radiotherapy. Radiat. Oncol. 2011, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schnarr, K.; Boreham, D.; Sathya, J.; Julian, J.; Dayes, I.S. Radiation-induced lymphocyte apoptosis to predict radiation therapy late toxicity in prostate cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Foro, P.; Algara, M.; Lozano, J.; Rodriguez, N.; Sanz, X.; Torres, E.; Carles, J.; Reig, A.; Membrive, I.; Quera, J.; et al. Relationship between radiation-induced apoptosis of t lymphocytes and chronic toxicity in patients with prostate cancer treated by radiation therapy: A prospective study. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Pouliliou, S.E.; Lialiaris, T.S.; Dimitriou, T.; Giatromanolaki, A.; Papazoglou, D.; Pappa, A.; Pistevou, K.; Kalamida, D.; Koukourakis, M.I. Survival Fraction at 2 Gy and γH2AX Expression Kinetics in Peripheral Blood Lymphocytes From Cancer Patients: Relationship With Acute Radiation-Induced Toxicities. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, P.A. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2009, 47, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.; Bouffler, S.; Mullenders, H.F.L.; Paretzke, H.; Sabatier, L. RISC-RAD radiosensitivity of individuals and susceptibility to cancer induced by ionizing radiations. Radioprotection 2008, 43, 238. [Google Scholar] [CrossRef]
- Akulevich, N.M.; Saenko, V.A.; Rogounovitch, T.I.; Drozd, V.M.; Lushnikov, E.F.; Ivanov, V.K.; Mitsutake, N.; Kominami, R.; Yamashita, S. Polymorphisms of DNA damage response genes in radiation-related and sporadic papillary thyroid carcinoma. Endocr. Relat. Cancer 2009, 16, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Nakachi, K.; Hayashi, T.; Hamatani, K.; Eguchi, H.; Kusunoki, Y. Sixty years of follow-up of Hiroshima and Nagasaki survivors: Current progress in molecular epidemiology studies. Mutat. Res. 2008, 659, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, A.; Rössler, U.; Hornhardt, S.; Sauter, W.; Bickeböller, H.; Wichmann, H.E.; Gomolka, M. Heritability of radiation response in lung cancer families. Genes 2012, 3, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Fowble, B.; Hanlon, A.; Freedman, G.; Nicolaou, N.; Anderson, P. Second cancers after conservative surgery and radiation for stages I–II breast cancer: Identifying a subset of women at increased risk. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 679–690. [Google Scholar] [CrossRef]
- Hosking, F.J.; Feldman, D.; Bruchim, R.; Olver, B.; Lloyd, A.; Vijayakrishnan, J.; Flint-Richter, P.; Broderick, P.; Houlston, R.S.; Sadetzki, S. Search for inherited susceptibility to radiation-associated meningioma by genomewide SNP linkage disequilibrium mapping. Br. J. Cancer 2011, 104, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Jansen-van der Weide, M.C.; Greuter, M.J.W.; Jansen, L.; Oosterwijk, J.C.; Pijnappel, R.M.; de Bock, G.H. Exposure to low-dose radiation and the risk of breast cancer among women with a familial or genetic predisposition: A meta-analysis. Eur. Radiol. 2010, 20, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Heymann, S.; Delaloge, S.; Rahal, A.; Caron, O.; Frebourg, T.; Barreau, L.; Pachet, C.; Mathieu, M.-C.; Marsiglia, H.; Bourgier, C. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat. Oncol. 2010, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- John, E.M.; Phipps, A.I.; Knight, J.A.; Milne, R.L.; Dite, G.S.; Hopper, J.L.; Andrulis, I.L.; Southey, M.; Giles, G.G.; West, D.W.; et al. Medical radiation exposure and breast cancer risk: Findings from the breast cancer family registry. Int. J. Cancer 2007, 121, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Elsakov, P.; Kurtinaitis, J.; Ostapenko, V. Prognostic value of BRCA1 mutations in familial breast cancer patients affected by a second primary cancer. Fam. Cancer 2007, 6, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Soto, G.; Ortiz-López, R.; Rojas-Martínez, A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet. Mol. Biol. 2015, 38, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.N.; Ingham, S.; Hodson, J.; Lalloo, F.; Bulman, M.; Howell, A.; Evans, D.G. Risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: A 30-year semi-prospective analysis. Fam. Cancer 2015, 14, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Best, T.; Li, D.L.; Skol, A.D.; Kirchhoff, T.; Jackson, S.A.; Yasui, Y.; Bhatia, S.; Strong, L.C.; Domchek, S.M.; Nathanson, K.L.; et al. Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin’s lymphoma. Nat. Med. 2011, 17, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Roberson, J.D.; Burnett, O.L.; Robin, N. Radiogenomics: Towards a personalized radiation oncology. Curr. Opin. Pediatr. 2016, 28, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Burrill, W.; Barber, J.B.; Roberts, S.A.; Bulman, B.; Scott, D. Heritability of chromosomal radiosensitivity in breast cancer patients: A pilot study with the lymphocyte micronucleus assay. Int. J. Radiat. Biol. 2009, 76, 1617–1619. [Google Scholar] [CrossRef]
- Docherty, Z.; Georgiou, A.; Langman, C.; Kesterton, I.; Rose, S.; Camplejohn, R.; Ball, J.; Barwell, J.; Gilchrist, R.; Pangon, L.; et al. Is chromosome radiosensitivity and apoptotic response to irradiation correlated with cancer susceptibility? Int. J. Radiat. Biol. 2009, 83, 1–12. [Google Scholar] [CrossRef]
- Kato, T.A.; Wilson, P.F.; Nagasawa, H.; Fitzek, M.M.; Weil, M.M.; Little, J.B.; Bedford, J.S. A defect in DNA double strand break processing in cells from unaffected parents of retinoblastoma patients and other apparently normal humans. DNA Repair 2007, 6, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.J.; Brock, W.A.; Chapman, J.D.; Wilson, G.; Fowler, J.F. Response predictors in radiotherapy: A review of research into radiobiologically based assays. Br. J. Radiol. Suppl. 1988, 22, 96–108. [Google Scholar] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γ-H2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Gheorghiu, L.; Liu, Q.; Efstathiou, J.A.; Wirth, L.J.; Krause, M.; von Neubeck, C. DNA damage response assessments in human tumor samples provide functional biomarkers of radiosensitivity. Semin. Radiat. Oncol. 2015, 25, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Martin, O.A.; Parekh, P.R.; Weyemi, U.S.; Bonner, W.M. Recent developments in the use of γ-H2AX as a quantitative DNA double-strand break biomarker. Aging 2011, 3, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.L.; Breton, J.; Douki, T.; Gasparutto, D.; Grand, A.; Rachidi, W.; Sauvaigo, S. Radiation-mediated formation of complex damage to DNA: A chemical aspect overview. Br. J. Radiol. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, I.P.; Golubov, A.; Koturbash, I.V.; Kutanzi, K.; Martin, O.A.; Kovalchuk, O. Age-dependent changes in DNA repair in radiation-exposed mice. Radiat. Res. 2014, 182, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. Irradiation induced foci (IRIF) as a biomarker for radiosensitivity. Mutat. Res. 2012, 736, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Bohgaki, T.; Bohgaki, M.; Hakem, R. DNA double-strand break signaling and human disorders. Genome Integr. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Fricke, A.; Schneider, R.; Simon, K.; Kuhne, M.; Fleckenstein, J.; Graber, S.; Graf, N.; Rube, C. DNA repair alterations in children with pediatric malignancies: Novel opportunities to identify patients at risk for high-grade toxicities. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Schuler, N.; Palm, J.; Kaiser, M.; Betten, D.; Furtwangler, R.; Rube, C.; Graf, N.; Rube, C.E. DNA-damage foci to detect and characterize DNA repair alterations in children treated for pediatric malignancies. PLoS ONE 2014, 9, e91319. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.L.K.; Horn, S.; Somaiah, N.; Davies, S.; Gothard, L.; A’Hern, R.; Yarnold, J.; Rothkamm, K. DNA double-strand break repair and induction of apoptosis in ex vivo irradiated blood lymphocytes in relation to late normal tissue reactions following breast radiotherapy. Radiat. Environ. Biophys. 2014, 53, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.L.K.; Somaiah, N.; A’Hern, R.; Davies, S.; Gothard, L.; Yarnold, J.; Rothkamm, K. Residual DNA and chromosomal damage in ex vivo irradiated blood lymphocytes correlated with late normal tissue response to breast radiotherapy. Radiother. Oncol. 2011, 99, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Mumbrekar, K.D.; Fernandes, D.J.; Goutham, H.V.; Sharan, K.; Vadhiraja, B.M.; Satyamoorthy, K.; Sadashiva, S.R.B. Influence of double-strand break repair on radiation therapy-induced acute skin reactions in breast cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Goutham, H.V.; Mumbrekar, K.D.; Vadhiraja, B.M.; Fernandes, D.J.; Sharan, K.; Parashiva, G.K.; Kapaettu, S.; Sadashiva, S.R.B. DNA double-strand break analysis by γ-H2AX foci: A useful method for determining the overreactors to radiation-induced acute reactions among head-and-neck cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, E607–E612. [Google Scholar] [CrossRef] [PubMed]
- Einwallner, E.; Subasic, A.; Strasser, A.; Augustin, D.; Thalhammer, R.; Steiner, I.; Schwarzinger, I. Lysis matters: Red cell lysis with FACS Lyse affects the flow cytometric enumeration of circulating leukemic blasts. J. Immunol. Methods 2013, 390, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, K.; Pinkawa, M.; Eble, M.J.; Muller, W.U.; Wojcik, A.; Kriehuber, R.; Schmitz, S. In vivo versus in vitro individual radiosensitivity analysed in healthy donors and in prostate cancer patients with and without severe side effects after radiotherapy. Int. J. Radiat. Biol. 2012, 88, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Van Oorschot, B.; Hovingh, S.E.; Moerland, P.D.; Medema, J.P.; Stalpers, L.J.A.; Vrieling, H.; Franken, N.A.P. Reduced activity of double-strand break repair genes in prostate cancer patients with late normal tissue radiation toxicity. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.; Martin, O.A.; Roos, D.E.; Lobachevsky, P.N.; Potter, A.E.; Zacest, A.C.; Bezak, E.; Bonner, W.M.; Martin, R.F.; Leong, T. Enhanced intrinsic radiosensitivity after treatment with stereotactic radiosurgery for an acoustic neuroma. Radiother. Oncol. 2012, 103, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.V.; Demidenko, Z.N.; Bukreeva, E.I.; Pospelov, V.A.; Gudkov, A.V.; Blagosklonny, M.V. Pseudo-DNA damage response in senescent cells. Cell Cycle 2009, 8, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P.; Keyes, M. Residual γH2AX after irradiation of human lymphocytes and monocytes in vitro and its relation to late effects after prostate brachytherapy. Radiother. Oncol. 2008, 86, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Vasireddy, R.S.; Sprung, C.N.; Cempaka, N.L.; Chao, M.; McKay, M.J. H2AX phosphorylation screen of cells from radiosensitive cancer patients reveals a novel DNA double-strand break repair cellular phenotype. Br. J. Cancer 2010, 102, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Du, C.-R.; Xu, W.-C.; Shi, Z.-L.; Zhang, Q.; Li, Z.-B.; Fu, S. Correlation of dynamic changes in γ-H2AX expression in peripheral blood lymphocytes from head and neck cancer patients with radiation-induced oral mucositis. Radiat. Oncol. 2013, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bourton, E.C.; Plowman, P.N.; Smith, D.; Arlett, C.F.; Parris, C.N. Prolonged expression of the γ-H2AX DNA repair biomarker correlates with excess acute and chronic toxicity from radiotherapy treatment. Int. J. Cancer 2011, 129, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, J.; Kühne, M.; Seegmüller, K.; Derschang, S.; Melchior, P.; Gräber, S.; Fricke, A.; Rübe, C.E.; Rübe, C. The impact of individual in vivo repair of DNA double-strand breaks on oral mucositis in adjuvant radiotherapy of head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Nahas, S.A.; Tunuguntla, R.; Fike, F.; Gatti, R.A. Assessing ‘radiosensitivity’ with kinetic profiles of γ-H2AX, 53BP1 and BRCA1 foci. Radiother. Oncol. 2011, 101, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Werbrouck, J.; Duprez, F.; De Neve, W.; Thierens, H. Lack of a correlation between γH2AX foci kinetics in lymphocytes and the severity of acute normal tissue reactions during IMRT treatment for head and neck cancer. Int. J. Radiat. Biol. 2011, 87, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Lobachevsky, P.; Leong, T.; Daly, P.; Smith, J.; Best, N.; Tomaszewski, J.; Thompson, E.R.; Li, N.; Campbell, I.G.; Martin, R.F.; et al. Compromized DNA repair as a basis for identification of cancer radiotherapy patients with extreme radiosensitivity. Cancer Lett. 2016, 383, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, C.N.; Alsner, J. Genetic variants and normal tissue toxicity after radiotherapy: A systematic review. Radiother. Oncol. 2009, 92, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.L.M.; Shihabuddeen, W.A.; Shwe, T.T.; Tan, S.H.; Wee, J.; Tan, T. Lymphocyte apoptosis as a predictive biomarker for radiotherapy de-intensification in EBV-associated nasopharynx cancer. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Braunstein, S.; Nakamura, J.L. Radiotherapy-induced malignancies: Review of clinical features, pathobiology, and evolving approaches for mitigating risk. Front. Oncol. 2013, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Berrington de Gonzalez, A.; Gilbert, E.; Curtis, R.; Inskip, P.; Kleinerman, R.; Morton, L.; Rajaraman, P.; Little, M.P. Second solid cancers after radiation therapy: A systematic review of the epidemiologic studies of the radiation dose-response relationship. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Kamran, S.C.; de Gonzalez, A.; Ng, A.; Haas-Kogan, D.; Viswanathan, A.N. Therapeutic radiation and the potential risk of second malignancies. Cancer 2016, 122, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Barnard, S.; Rothkamm, K. γ-H2AX-based dose estimation for whole and partial body radiation exposure. PLoS ONE 2011, 6, e25113. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Yin, X.; Forrester, H.B.; Sprung, C.N.; Martin, R.F. Potential strategies to ameliorate risk of radiotherapy-induced second malignant neoplasms. Semin. Cancer Biol. 2016, 37, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Forker, L.J.; Choudhury, A.; Kiltie, A.E. Biomarkers of tumour radiosensitivity and predicting benefit from radiotherapy. Clin. Oncol. 2015, 27, 561–569. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habash, M.; Bohorquez, L.C.; Kyriakou, E.; Kron, T.; Martin, O.A.; Blyth, B.J. Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer. Cancers 2017, 9, 147. https://doi.org/10.3390/cancers9110147
Habash M, Bohorquez LC, Kyriakou E, Kron T, Martin OA, Blyth BJ. Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer. Cancers. 2017; 9(11):147. https://doi.org/10.3390/cancers9110147
Chicago/Turabian StyleHabash, Mohammad, Luis C. Bohorquez, Elizabeth Kyriakou, Tomas Kron, Olga A. Martin, and Benjamin J. Blyth. 2017. "Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer" Cancers 9, no. 11: 147. https://doi.org/10.3390/cancers9110147
APA StyleHabash, M., Bohorquez, L. C., Kyriakou, E., Kron, T., Martin, O. A., & Blyth, B. J. (2017). Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer. Cancers, 9(11), 147. https://doi.org/10.3390/cancers9110147