1. Introduction
Advancements in cancer biology have led to improvements in cancer mortality rates; however, therapeutic resistance continues to be a major cause of treatment failure in cancer. In particular, glioblastoma multiforme (GBM) is an aggressive type of brain cancer with poor prognosis. If GBM patients do not succumb to their original tumor, despite surgical resection and post-operative radiation and chemotherapy, the tumor nearly always recurs. The failure in treating GBM has largely been attributed to heterogeneity within the tumor cell population [
1,
2]. Oftentimes, these front-line cancer treatments successfully eliminate the bulk of the tumor formed by fast proliferating GBM cells, namely the tumor mass-forming cells (TMC), yet it spares the invasive GBM cells, namely the stem-like tumor-initiating cells (STIC). This results in an increase in the treatment-resistant population. After treatment, chromosome instability may restore intra-tumoral heterogeneity in the surviving population and enable the continued evolution of the tumor, leading to recurrence.
In addition to different responses to front-line cancer therapies, heterogeneity in the molecular profile of the subpopulations also results in different responses to targeted therapies between the subpopulations. The epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase that conducts one of the few evolutionarily-conserved signaling pathways. This pathway dictates many aspects of embryonic and post-embryonic development in multicellular organisms, and re-activation of this signaling pathway has been associated with cancer. Thus, cancer has long been viewed as a disease of cellular de-differentiation. EGFR is commonly overexpressed or activated via gene amplified and/or gain of function mutations in a variety of cancers. EGFR triggers AKT-centered oncogenic signaling pathways, and high EGFR levels also signify a high state of cancer cellular respiration. In GBM, nearly 60% of patients have abnormalities on EGFR [
3], rendering EGFR as a compelling drug target. However, intra-tumoral heterogeneity produces functional subpopulations, including STIC and TMC, with different levels of EGFR expression, and decades-long trials with anti-EGFR agents have failed to improved GBM patient outcomes [
4]. The challenge lies in identifying subsets of the tumor with molecular profiles which may benefit from these specific targeted therapies. Therefore, the ability to monitor transitions in functional tumor subpopulations and track their responses to treatment will lead to a better understanding of tumor progression and drug resistance.
Early studies have distinguished functional subpopulations by cell surface markers, including CD133 for brain tumor initiating cells [
5]. The identification of surface markers for specific subpopulations has confirmed intra-tumoral heterogeneity; however, limitations of the methods for in vitro identification of cell surface markers prevent its application to tracking dynamic population shifts in vivo. Therefore, recent studies have applied non-invasive and label-free metabolic imaging methods to observe mixtures of tumor populations [
6].
Techniques to monitor cancer progression have commonly exploited the altered metabolism of cancer relative to normal tissue. The preference for glycolysis over oxidative phosphorylation (OXPHOS) in neoplastic tissue is known as the Warburg effect [
7,
8]. The increased uptake of glucose and a structural analogue,
18F-fluorodeoxyglucose, by cancer due to its altered metabolism has been exploited as a diagnostic tool, known as
18F-fluorodeoxyglucose-positron emission tomography (
18F-FDG-PET), to accurately predict histological grading and survival in patients with gliomas [
9]. In addition to differences in metabolism between normal and neoplastic tissues, metabolic differences exist between different tumor subpopulations. Our previous work applied the Seahorse XF assay, which directly quantifies OXPHOS through the measurement of oxygen consumption rate (OCR), and quantifies glycolytic respiration through the measurement of extracellular acidification rate (ECAR) in the culture media surrounding the cells. Subsequent challenges to the cells are then applied through metabolic inhibitors to reveal the reserved (spare) respiration as a response to stress. In our application of the Seahorse assay, STIC-enriched GBM lines consistently had lower basal OCR and a minimal ability for OXPHOS to respond to stress, relative to their syngeneic TMC enriched line [
10,
11]. Due to these metabolic differences, we propose to apply metabolic imaging methods to non-invasively track tumor subpopulations.
Metabolic imaging methods utilize the fluorescence of the endogenous metabolic coenzymes reduced nicotinamide dinucleotide (NADH) and flavin adenine dinucleotide (FAD) to non-invasively probe the metabolic state of the cell without the use of exogenous labels. Fluorescence lifetime imaging microscopy (FLIM) of protein-bound and free-NADH has previously detected an increase in the fraction of free NADH with an increase in the neoplastic metabolism in a hamster cheek pouch model [
12]. In addition, an increase in the fraction of free NADH was also detected in highly proliferative stem cells at the base of the intestinal crypt relative to the nearby differentiated cells of the small intestinal epithelia [
13]. These studies suggest an increase in the fraction of free NADH correlates with an increase in glycolysis and cell proliferation. In contrast to the Seahorse assay, which is applied to cells in vitro, these studies demonstrate FLIM’s ability to be applied both in vitro and in vivo. Furthermore, the spatial resolution provided by metabolic imaging techniques enable the measurement of cell metabolism of individual cells, whereas the Seahorse assay measures bulk behaviors of entire populations. The ability to measure cells individually allows for the classification into specific subpopulations. In recent studies, this has been demonstrated in a co-culture model to identify a mixture of two different breast cancer cell lines [
6]. It has also been applied to distinguish proliferating, quiescent and apoptotic cells in an acute myeloid leukemia model [
14]. These studies reinforce the value of optical metabolic imaging methods in classifying tumor subpopulations.
FLIM’s ability to measure cellular metabolic states and provide spatial resolution makes it an ideal tool for studying intra-tumoral heterogeneity. In this study, the phasor approach to FLIM analysis of NADH was applied to distinguish and track functional tumor subpopulations of GBM, specifically the invasive STIC and the proliferative TMC subpopulations. The two subpopulations were enriched using established cell culture conditions [
2]. The functions of both of these lines were previously verified in vivo [
11,
15]. After distinguishing the two subpopulations in vitro and in vivo by FLIM, we monitored the transition from a STIC dominant to a TMC dominant subpopulation. These results validated FLIM as a sensitive and non-invasive method to track tumor subpopulation dynamics with spatial resolution to potentially identify tumor cell behavior during treatment.
3. Discussion
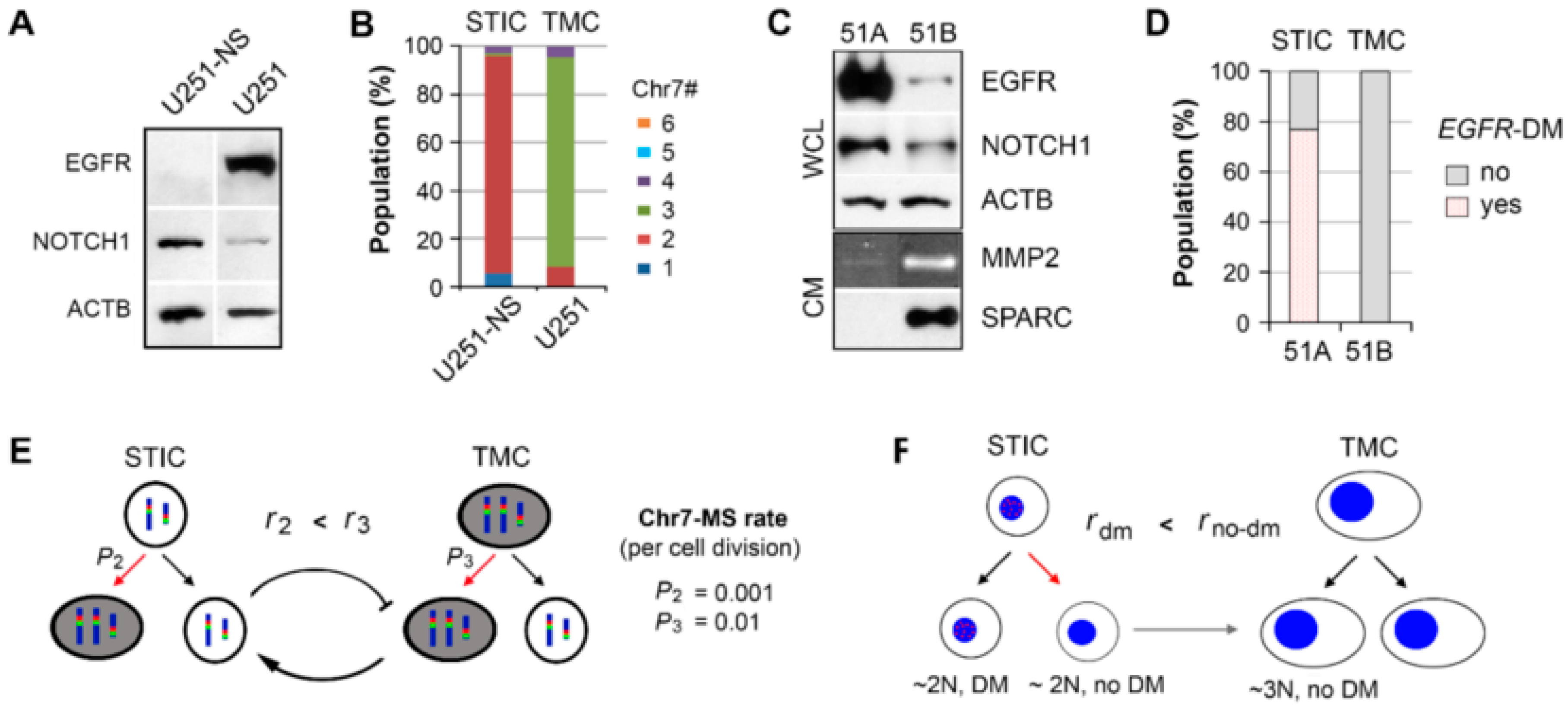
In this study, two syngeneic cultures containing subpopulations of cells functionally characterized in vivo to be fast growing (TMC) and highly invasive (STIC) were imaged and monitored by phasor FLIM of NADH. Enrichment for the two subpopulations was achieved by SA and NS-culture conditions for TMC and STIC populations, respectively. This study included the TMC-enriched U251 and STIC-enriched U251-NS syngeneic GBM cell lines and the TMC-enriched 51B and STIC-enriched 51A syngeneic GBM primary cultures. Although U251 and 51B were functionally similar in vivo relative to U251-NS and 51A in their contributions to tumor growth and invasive behaviors, these cultures were found to differ in oncogenic protein/gene expressions and karyotypes (
Figure 1). However, due to their similarities in high invasiveness versus high growth rate, we hypothesized that FLIM of NADH could discriminate between these two tumor subpopulations and monitor the transition from STIC to TMC, which underlies tumor recurrence.
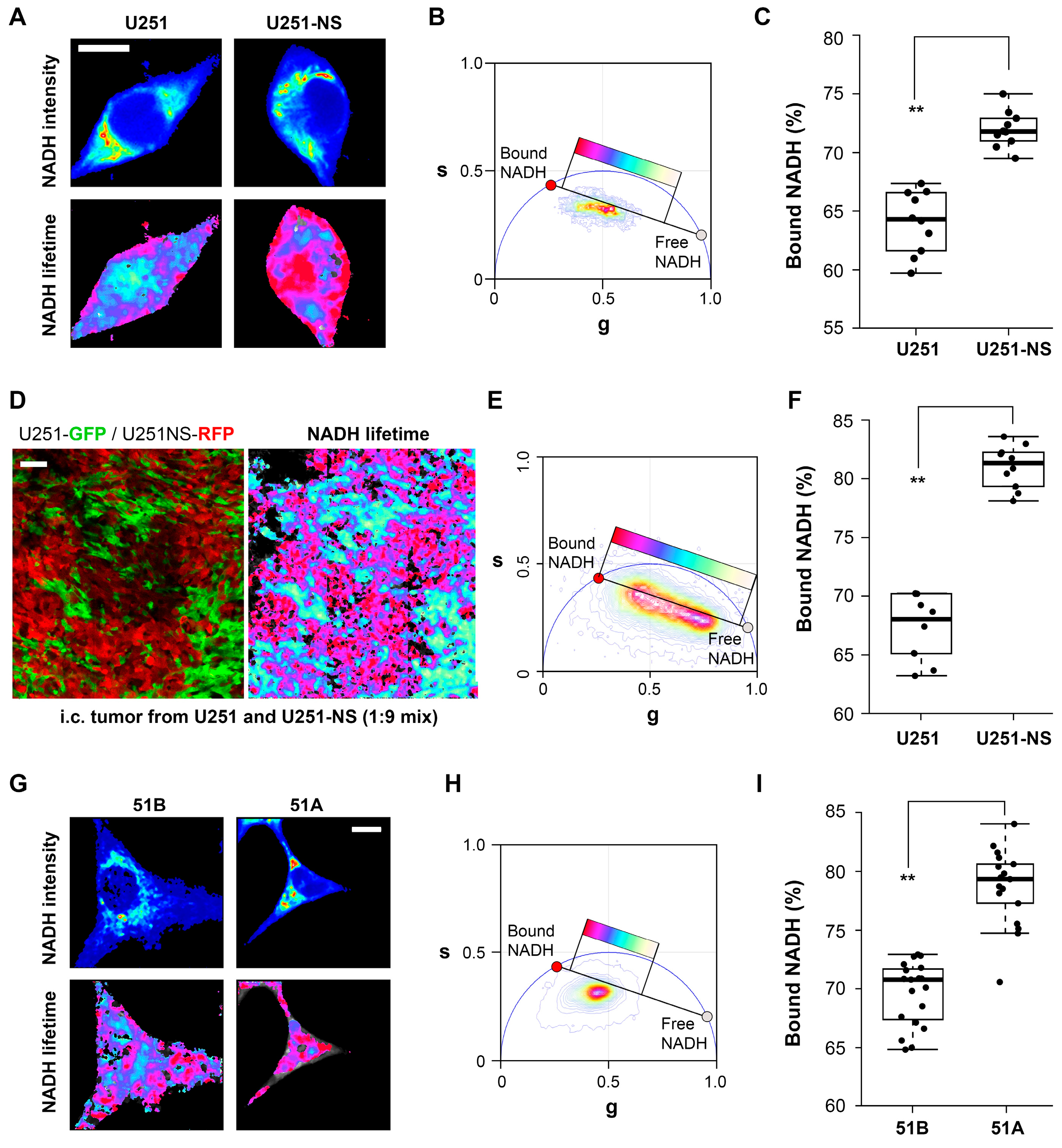
When imaged in vitro, FLIM identified a higher fraction of bound NADH in both of the STIC-enriched cultures, U251-NS and 51A, relative to their TMC-enriched cultures, U251 and 51B (
Figure 2). This suggests that the functional difference between the TMC and STIC subpopulations can be monitored by the changes in the protein bound or free fractions of NADH by phasor FLIM. To confirm that the distinct phasor FLIM signatures are related to the subpopulation and not an artifact of in vitro culture conditions, the enriched populations were fluorescently labeled and implanted as intracranial xenografts in a mouse model. In agreement with the in vitro results, the in vivo results further supported the ability for phasor FLIM of NADH to distinguish STIC and TMC subpopulations.
An increase in the fraction of free NADH measured by FLIM has been reported to be associated with a transition from OXPHOS to glycolysis and an increase in cell proliferation in cancer development [
12,
18]. Therefore, it was partially unexpected that a high fraction of bound NADH was identified by FLIM in the STIC in both GBM models, where the Seahorse assay measured lower OXPHOS in both cultures, relative to each respective TMC-enriched culture [
10,
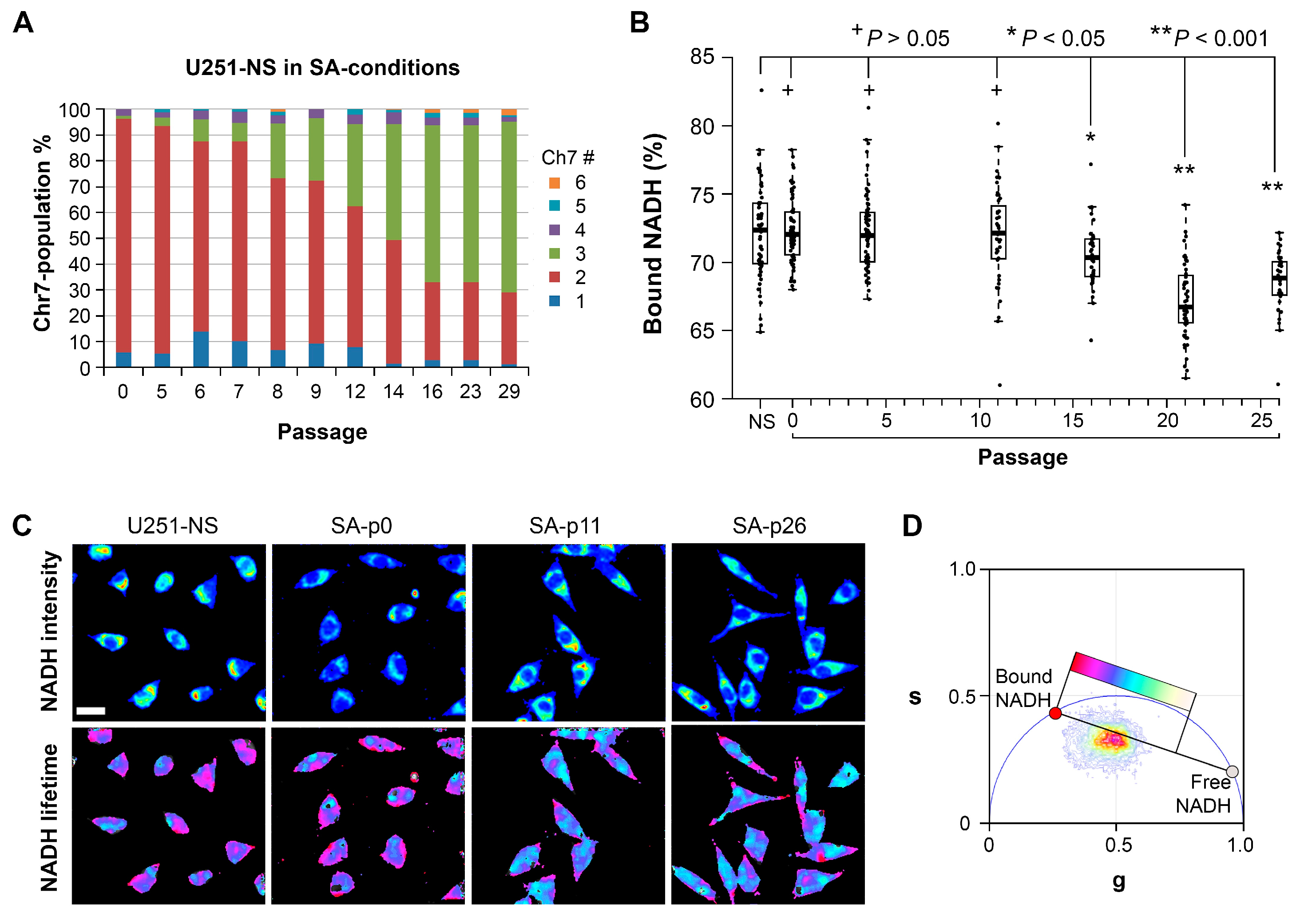
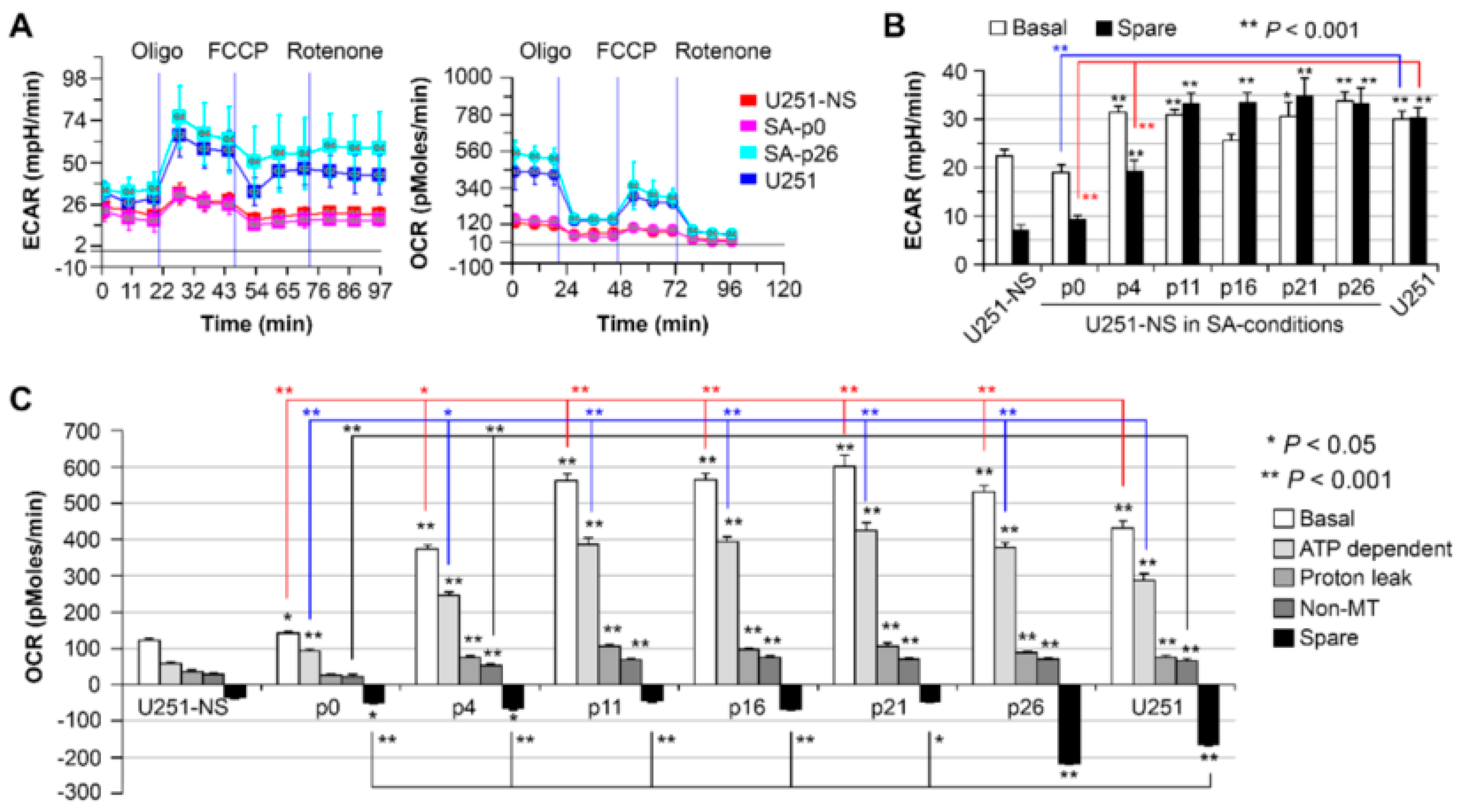
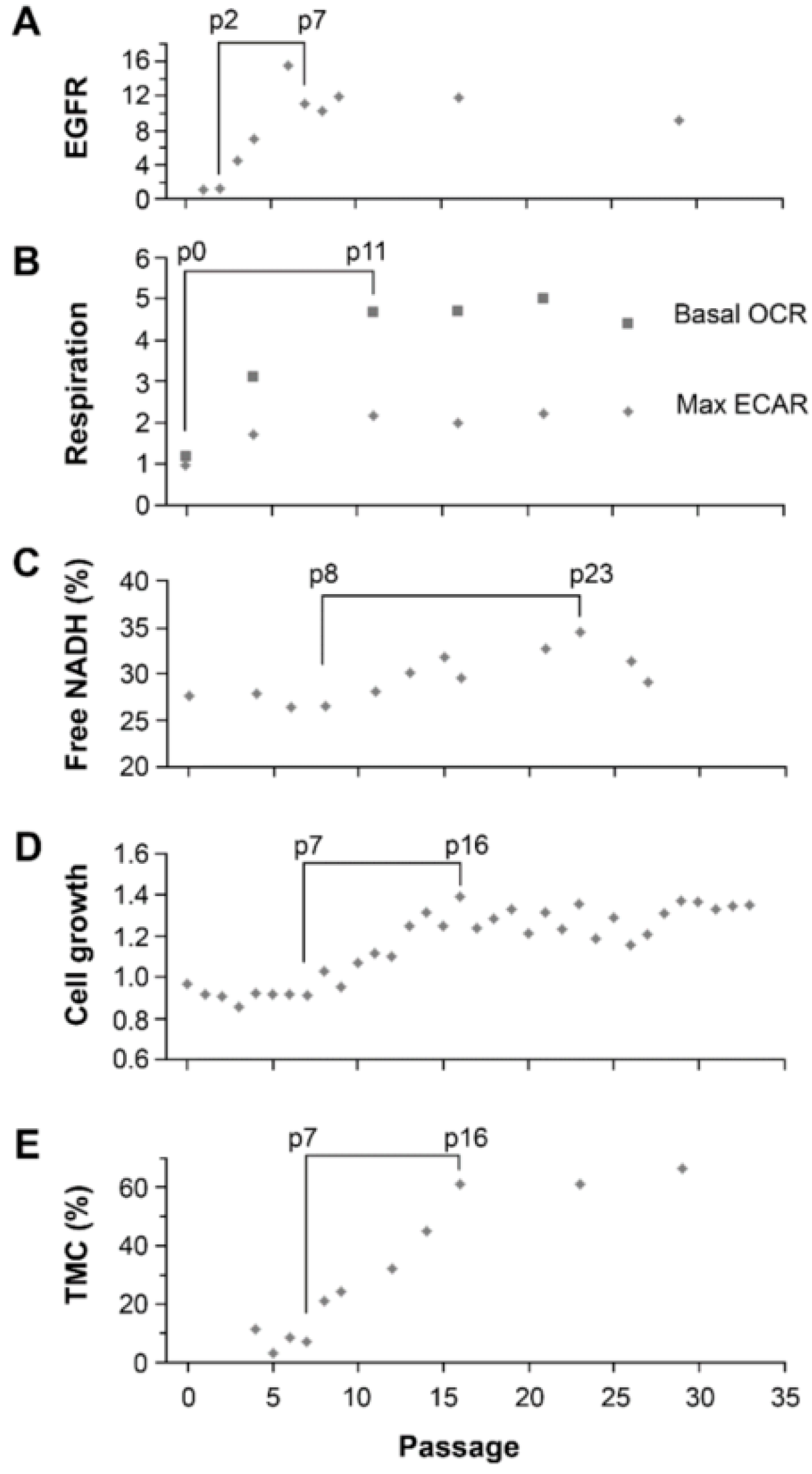
11]. However, the increase in the fraction of bound-NADH does correlate as expected to the decrease in proliferation rates in the STICS population relative to the TMC population. Results from our in vitro model, which changed the ratio of STIC and TMC, showed increases of both OXPHOS and glycolysis in response to environmental changes occurred before an increase in TMC fraction, an increase of the free NADH fraction, and an increase in cell growth rate (
Figure 6). Hence, increases in cell respiration of both OXPHOS and glycolysis in STIC cells are likely related to activation of EGFR signaling, but not related to subpopulation identity.
Previous studies applying FLIM of NADH have primarily relied on relative changes within a single population of cells to infer metabolic shifts. Our studies used syngeneic cell lines from two GBMs, differentially enriched with two functional subpopulations (TMC and STIC). To our knowledge, this is the first study to show distinctly different baseline fractions of bound-NADH in functionally validated tumor cells (STIC and TMC). Additional work will be needed to further understand the link between the high fraction of bound NADH in STIC and their low OXPHOS metabolic state in malignant gliomas.
Although the mechanism by which STIC of malignant gliomas maintain a high fraction of bound NADH is unclear, our model of changing the majority of cells from STIC in U251-NS into TMC as seen in U251 by switching the culture from NS to SA-conditions revealed a strong correlation in time between the increase in fraction of free NADH by FLIM and the increase of cell growth rate (rs = 0.86, p = 0.01), which supports previous findings of a positive relationship between these two variables. In addition, a strong correlation also exists between the fraction of bound NADH and percentage of TMC (rs = 0.80), although the significance is low due to the sparsity of measurements (p = 0.20, only four measurements for NADH data during the included passage numbers).
The ability to monitor changes in tumor subpopulations would be highly beneficial to an understanding of drug resistance. To our knowledge, this study is the first to apply FLIM to identify functional tumor subpopulations of the same tumor origin. This study demonstrates that FLIM can identify functionally similar subpopulations in different GBMs that apparently have undergone different paths of cancer evolution. Specifically, it has identified that the subpopulation of the STIC phenotype has a higher fraction of bound NADH relative to the subpopulation of the TMC phenotype. Furthermore, phasor FLIM of NADH is able to dynamically track transitions between these two subpopulations over time. Overall, this suggests that the NADH state measured by FLIM will become a valuable tool for understanding the changes in functional subpopulations during cancer treatment.
4. Materials and Methods
4.1. Cell Cultures and Protein Analysis
Human GBM-derived cell line U251, its lentiviral pGIPZ-transductant U251-GFP, its clonal subculture U251-NS and lentiviral pTRIPZ-transductant U251NS-RFP were described in Hu et al., 2013 [
15]. Human GBM-derived syngeneic primary cultures of 51A and 51B were established at the UC Irvine Brain Tumor Research Lab described in Zhou et al., 2017 [
11]. Cells used in this study were authenticated and validated to be free of mycoplasma using MycoAlert™ PLUS Mycoplasma Detection Kit (Lonza, Walkersville, MD, USA).
U251, U251-GFP and 51B were cultured in serum-containing (5% FBS for U251 and U251-GFP, 10% FBS for 51B) DMEM/F12 medium under adherent (SA) conditions. U251-NS, U251NS-RFP and 51A were cultured in neurosphere conditions (NS) in DMEM/F12 supplemented with epidermal growth factor (EGF, 20 ng/mL), basic fibroblast growth factor (FGF, 10 ng/mL) and 1% B27 (Invitrogen, Carlsbad, CA, USA). Prior to the study, monolayer cultures of U251-NS, U251NS-RFP and 51A were achieved by culture in fibronectin-coated dishes (1 μg/cm2). All cells were cultured in a humidity chamber containing 5% CO2 at 37 °C.
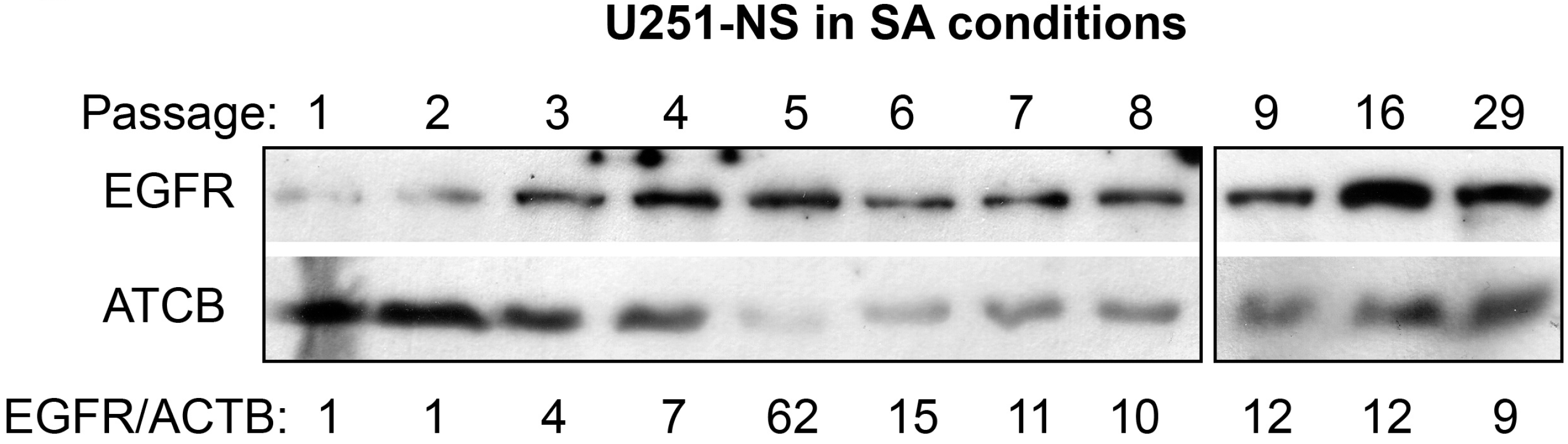
Protein analyses were carried out for U251-NS cultured and serially passed in SA-conditions, for the expression of EGFR. Primary antibodies for EGFR (rabbit mAb, Cell Signaling Technology, Inc., Danvers, MA, USA) and ACTB (IgM-specific mouse, Millipore, Burlington, MA, USA) were diluted 1:10,000 for immunoblotting as described previously [
19].
4.2. Intracranial Xenografts
The animal work was approved by the Animal Care and Use Committee (IACUC) of the University of California, Irvine. The establishment of intracranial (i.c.) xenografts from glioma cells was described previously [
15]. Briefly, U251-GFP and U251-RFP cells were mixed in a 1:9 ratio and injected (1 × 10
5 cells/3 μL DMEM/F12) into the frontal lobe of 4–6-week-old, female, nude mice (strain NCrNu-M, Taconic, Hudson, NY, USA). After i.c. implantation, mice were periodically observed for moribund signs (hunchback posture, marked weight loss, and gait impairment). Any mouse showing 15–20% body weight loss was euthanized, and its brain was immediately removed and placed in a mouse brain slicer. 1mm think slices of mouse brain containing tumor cells (verified under a fluorescence microscope for RFP and GFP-expressing tumor cells) were placed in a 4-well chamber slide containing 1 mL of DMEM/F12 medium for fluorescence lifetime imaging microscopy.
4.3. Seahorse XF-24 Extracellular Flux Analyses on Cellular Respiration
To measure respiration and mitochondrial function in glioma cells, we employed a Seahorse Bioscience XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) to directly measure the level of OXPHOS, by oxygen consumption rate (OCR), and glycolytic respiration, by the extracellular acidification rate (ECAR), and utilized sequential injections of oligomycin (a complex V inhibitor), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (a mitochondrial oxidative phosphorylation uncoupler) and Rotenone (a complex I inhibitor), to dissect components of OXPHOS and the capacity of maximum respiration for both OXPHOS and glycolysis. Cells were seeded into a non-coated (for U251 and U251-NS in culture of SA-conditions) or fibronectin-coated (for U251-NS in culture of NS-conditions) 24-well Seahorse XF-24 assay plate at plating densities of 50,000–150,000 cells/well according to their growth speed and cultured for 2 days prior to flux analyses, as described previously [
11].
4.4. Fluorescence Lifetime Imaging Microscopy
For in vitro FLIM analysis, cells were plated on fibronectin- or collagen-coated glass bottom imaging dishes 24 h prior to FLIM analysis. Data were acquired on a Zeiss LSM 710 (Carl Zeiss, Jena, Germany) microscope with incubation at 37 °C and 5% CO2 using a C-Apochromat 40×/1.2 N.A. water immersion objective (Carl Zeiss, Jena, Germany). For in vivo FLIM analysis, intracranial xenografts images were acquired on an Olympus FluoView FV1000 (Olympus, Tokyo, Japan) with incubation at 37 °C and 5% CO2 using a 40×/0.8 N.A. water immersion objective. Fluorescence confocal images were acquired followed by FLIM of NADH of the same area. To acquire a larger field of view, 3 × 3 images were acquired and stitched together to form a single image. For single-cell analysis of the in vivo images, individual cells were identified and masked by their fluorescent protein label of either GFP (505–540 nm emission) or RFP (575–620 nm emission). The average fraction of bound-NADH was then calculated for individual cells.
NADH was excited by an 80-MHz titanium:sapphire Mai Tai Laser (Spectra-Physics, Santa Clara, CA, USA) at 740 nm. Images were collected at a scan speed of 25.21 μs/pixel and a size of 256 × 256 pixels. For each fluorescence lifetime image, 50 frames were collected and integrated. The excitation was separated from the emission light by a 690-nm dichroic mirror, and the NADH emission was collected by a 460/80 bandpass filter and a photomultiplier tube (H7422P-40, Hamamatsu Photonics, Hamamatsu, Japan). Coumarin 6 in ethanol with a fluorescence lifetime of 2.5 ns was used as a standard to calibrate for the instrument’s response time. Frequency domain FLIM data were acquired by the A320 FastFLIM box (ISS, Champaign, IL, USA) and analyzed by SimFCS software (Laboratory for Fluorescence Dynamics, Irvine, CA, USA).
The phasor approach was applied to analyze fluorescence lifetime images as previously described [
20]. Briefly, every pixel of the integrated fluorescence lifetime image is transformed into a coordinate on the phasor plot. The g and s coordinates in the phasor plot are calculated from the sine and cosine components of the Fourier transform of the fluorescence intensity decay of each pixel in the image. In the phasor space, pixels with single exponential decays fall on the universal semicircle, while pixels with complex decays fall within the semicircle. If a pixel contains two molecular species, the phasor will appear along a straight line joining the phasors of the two species. The position on the line will depend on the relative fluorescence contribution of each species. When applied to NADH within cells, the phasor will fall on a line between the phasors of purely protein bound NADH (with a fluorescence lifetime of 3.4 ns) and purely free NADH (with a fluorescence lifetime of 0.4 ns) [
21]. From the position between the two phasors, the relative amounts of fluorescence from bound NADH and free NADH can be computed.
4.5. Statistical Analysis
The NS and SA-cultures were compared for differences in bound-NADH fraction, OCR and ECAR cellular respirations by 2-sample equal-variance t-tests with Excel 2010 (Microsoft Corp, Redmond, WA, USA). The dynamic changes in cell growth speed, EGFR protein level, fractions of trisomy-7 cell and free NADH in U251-NS over passages in SA-conditions (passage numbers 4–16, inclusive) were analyzed by Spearman correlation analysis with SAS Version 9.4 (The SAS Institute, Cary, NC, USA). Seahorse data were not included due to sparsity (fewer than 4 measurements) during the included passage numbers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





