
AR Signaling in Breast Cancer

Abstract
:1. Introduction
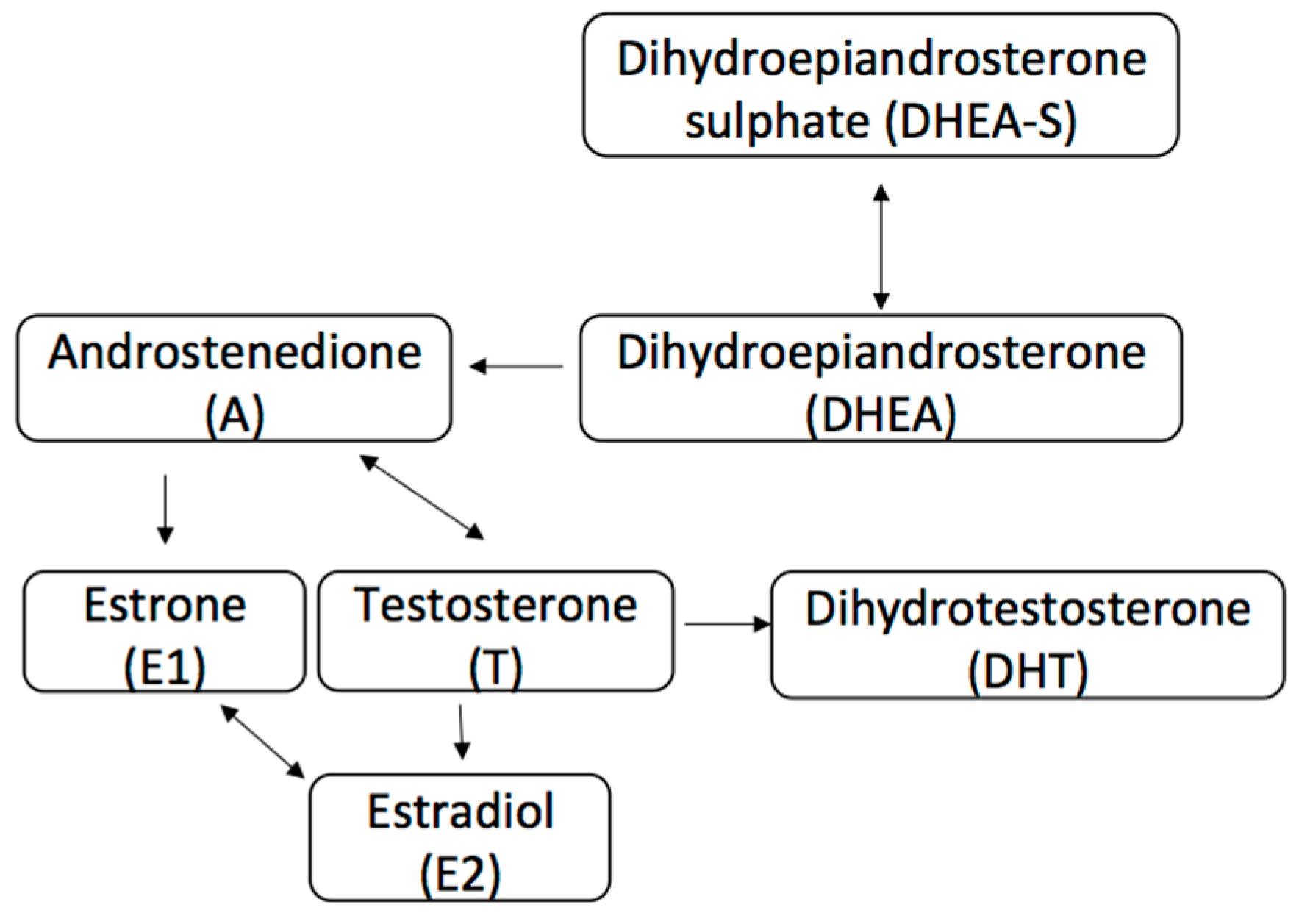
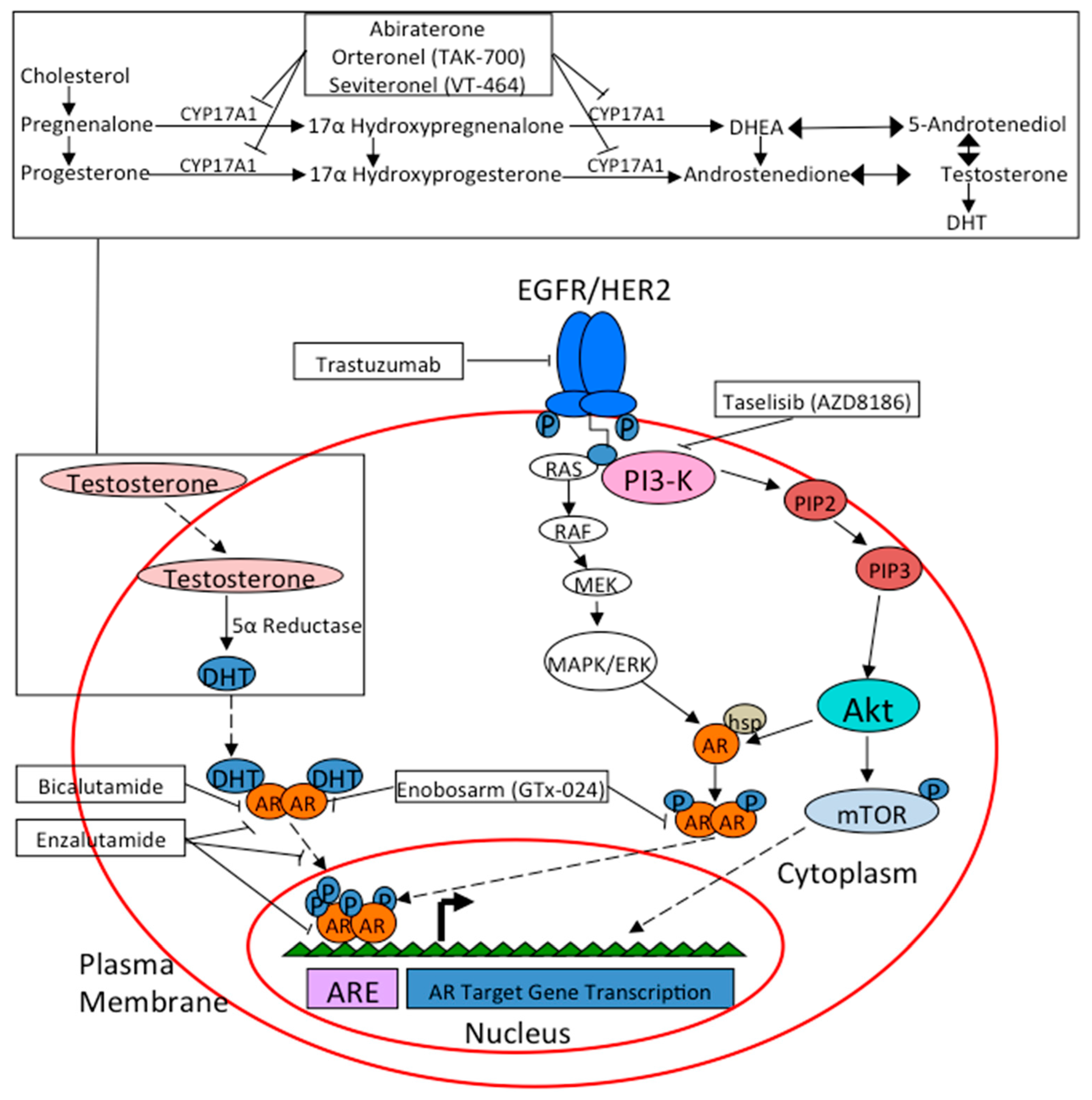
2. AR Pathway in Breast Cancer
3. AR Pathway in ER+ Breast Cancer
4. Prognostic Implications of AR in ER+ Breast Cancer
5. AR Pathway in HER2 Amplified Breast Cancer
6. Prognostic Implications of AR in HER2 Amplified Breast Cancer
7. AR Pathway in TNBC
8. Prognostic Implications of AR in TNBC Breast Cancer
9. Treatment Options in AR+ Breast Cancer
9.1. Bicalutamide
9.2. Enzalutamide
9.3. Abiraterone
9.4. Newer Anti-Androgens
9.5. SARMs
9.6. Other Drugs
10. Discussion
11. Conclusions
Conflicts of Interest
References
- Birrell, S.N.; Bentel, J.M.; Hickey, T.E.; Ricciardelli, C.; Weger, M.A.; Horsfall, D.J.; Tilley, W.D. Androgens induce divergent proliferative responses in human breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 1995, 52, 459–467. [Google Scholar] [CrossRef]
- Zhu, X.; Li, H.; Liu, J.P.; Funder, J.W. Androgen stimulates mitogen-activated protein kinase in human breast cancer cells. Mol. Cell. Endocrinol. 1999, 152, 199–206. [Google Scholar] [CrossRef]
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef] [PubMed]
- Foradori, C.D.; Weiser, M.J.; Handa, R.J. Non-genomic actions of androgens. Front. Neuroendocrinol. 2008, 29, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.G. Androgen production in women. Fertil. Steril. 2002, 77 (Suppl. S4), S3–S5. [Google Scholar] [CrossRef]
- Davison, S.L.; Davis, S.R. Androgens in women. J. Steroid Biochem. Mol. Biol. 2003, 85, 363–366. [Google Scholar] [CrossRef]
- Walters, K.A. Role of androgens in normal and pathological ovarian function. Reproduction 2015, 149, R193–R218. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Luu-The, V.; Labrie, C.; Belanger, A.; Simard, J.; Lin, S.X.; Pelletier, G. Endocrine and intracrine sources of androgens in women: Inhibition of breast cancer and other roles of androgens and their precursor dehydroepiandrosterone. Endocr. Rev. 2003, 24, 152–182. [Google Scholar] [CrossRef] [PubMed]
- Birrell, S.N.; Butler, L.M.; Harris, J.M.; Buchanan, G.; Tilley, W.D. Disruption of androgen receptor signaling by synthetic progestins may increase risk of developing breast cancer. FASEB J. 2007, 21, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.D.; Griffin, J.E.; Leshin, M.; George, F.W. Role of gonadal hormones in development of the sexual phenotypes. Hum. Genet. 1981, 58, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, A.M.; O’Neill, P.A.; Davies, M.P.; Sibson, R.; West, C.R.; Smith, P.H.; Foster, C.S. Declining estrogen receptor-beta expression defines malignant progression of human breast neoplasia. Am. J. Surg. Pathol. 2003, 27, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Skliris, G.P.; Munot, K.; Bell, S.M.; Carder, P.J.; Lane, S.; Horgan, K.; Lansdown, M.R.; Parkes, A.T.; Hanby, A.M.; Markham, A.F.; et al. Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J. Pathol. 2003, 201, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Roger, P.; Sahla, M.E.; Makela, S.; Gustafsson, J.A.; Baldet, P.; Rochefort, H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res. 2001, 61, 2537–2541. [Google Scholar] [PubMed]
- Marotti, J.D.; Collins, L.C.; Hu, R.; Tamimi, R.M. Estrogen receptor-beta expression in invasive breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod. Pathol. 2010, 23, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Cole, K.S.; Marotti, J.D.; Hu, R.; Schnitt, S.J.; Tamimi, R.M. Androgen receptor expression in breast cancer in relation to molecular phenotype: Results from the Nurses’ Health Study. Mod. Pathol. 2011, 24, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Niemeier, L.A.; Dabbs, D.J.; Beriwal, S.; Striebel, J.M.; Bhargava, R. Androgen receptor in breast cancer: Expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. 2010, 23, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Guedj, M.; Marisa, L.; de Reynies, A.; Orsetti, B.; Schiappa, R.; Bibeau, F.; MacGrogan, G.; Lerebours, F.; Finetti, P.; Longy, M.; et al. A refined molecular taxonomy of breast cancer. Oncogene 2012, 31, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Kuenen-Boumeester, V.; Van der Kwast, T.H.; Claassen, C.C.; Look, M.P.; Liem, G.S.; Klijn, J.G.; Henzen-Logmans, S.C. The clinical significance of androgen receptors in breast cancer and their relation to histological and cell biological parameters. Eur. J. Cancer 1996, 32, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Moinfar, F.; Okcu, M.; Tsybrovskyy, O.; Regitnig, P.; Lax, S.F.; Weybora, W.; Ratschek, M.; Tavassoli, F.A.; Denk, H. Androgen receptors frequently are expressed in breast carcinomas: Potential relevance to new therapeutic strategies. Cancer 2003, 98, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Vera-Badillo, F.E.; Templeton, A.J.; de Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen receptor expression and outcomes in early breast cancer: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef] [PubMed]
- McGhan, L.J.; McCullough, A.E.; Protheroe, C.A.; Dueck, A.C.; Lee, J.J.; Nunez-Nateras, R.; Castle, E.P.; Gray, R.J.; Wasif, N.; Goetz, M.P.; et al. Androgen receptor-positive triple negative breast cancer: A unique breast cancer subtype. Ann. Surg. Oncol. 2014, 21, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. Triple-negative breast cancer: Role of the androgen receptor. Cancer J. 2010, 16, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.H.; Choi, S.Y.; Lee, J.H.; Park, B.W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Chia, K.; O’Brien, M.; Brown, M.; Lim, E. Targeting the androgen receptor in breast cancer. Curr. Oncol. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.M.; Yoda, T.; Takagi, K.; Miki, Y.; Suzuki, T.; Sasano, H. Androgen receptor in triple negative breast cancer. J. Steroid Biochem. Mol. Biol. 2013, 133, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Fassan, M.; Cascione, L.; Guler, G.; Balci, S.; Irkkan, C.; Paisie, C.; Lovat, F.; Morrison, C.; Zhang, J.; et al. Androgen receptor status is a prognostic marker in non-basal triple negative breast cancers and determines novel therapeutic options. PLoS ONE 2014, 9, e88525. [Google Scholar] [CrossRef] [PubMed]
- Mrklic, I.; Pogorelic, Z.; Capkun, V.; Tomic, S. Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem. 2013, 115, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Chen, Y.; Lim, E.; Wimberly, H.; Bailey, S.T.; Imai, Y.; Rimm, D.L.; Liu, X.S.; Brown, M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 2011, 20, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.A.; El-Ayat, G.A. Non-operative breast pathology: Apocrine lesions. J. Clin. Pathol. 2007, 60, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Selim, A.G.; Wells, C.A. Immunohistochemical localisation of androgen receptor in apocrine metaplasia and apocrine adenosis of the breast: Relation to oestrogen and progesterone receptors. J. Clin. Pathol. 1999, 52, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, D.; Pakneshan, S.; Tavassoli, F.A. Androgen receptor (AR) expression in 400 breast carcinomas: Is routine AR assessment justified? Am. J. Cancer Res. 2014, 4, 353–368. [Google Scholar] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Ni, Y.B.; Chan, S.K.; Shao, M.M.; Law, B.K.; Tan, P.H.; Tse, G.M. Androgen receptor expression shows distinctive significance in ER positive and negative breast cancers. Ann. Surg. Oncol. 2014, 21, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.P.; Yang, Y.L.; Zhu, H.; Wang, J.; Jia, Y.; Liu, N.; Song, Y.J.; Zan, L.K.; Zhang, X.; Zhou, M.; et al. Expression of the androgen receptor and its correlation with molecular subtypes in 980 chinese breast cancer patients. Breast Cancer 2012, 6, 1–8. [Google Scholar] [PubMed]
- Cops, E.J.; Bianco-Miotto, T.; Moore, N.L.; Clarke, C.L.; Birrell, S.N.; Butler, L.M.; Tilley, W.D. Antiproliferative actions of the synthetic androgen, mibolerone, in breast cancer cells are mediated by both androgen and progesterone receptors. J. Steroid Biochem. Mol. Biol. 2008, 110, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, R.; Luttchens, S.; Hofmann, J.; Kunzmann, R.; Holzel, F.; Schulz, K.D. Androgen sensitivity of the new human breast cancer cell line MFM-223. Cancer Res. 1991, 51, 5722–5727. [Google Scholar] [PubMed]
- Poulin, R.; Baker, D.; Labrie, F. Androgens inhibit basal and estrogen-induced cell proliferation in the ZR-75-1 human breast cancer cell line. Breast Cancer Res. Treat. 1988, 12, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; De Amicis, F.; Rago, V.; Carpino, A.; Maggiolini, M.; Panno, M.L.; Lanzino, M. Breast cancer: From estrogen to androgen receptor. Mol. Cell. Endocrinol. 2002, 193, 121–128. [Google Scholar] [CrossRef]
- Need, E.F.; Selth, L.A.; Harris, T.J.; Birrell, S.N.; Tilley, W.D.; Buchanan, G. Research resource: Interplay between the genomic and transcriptional networks of androgen receptor and estrogen receptor alpha in luminal breast cancer cells. Mol. Endocrinol. 2012, 26, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Szelei, J.; Jimenez, J.; Soto, A.M.; Luizzi, M.F.; Sonnenschein, C. Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology 1997, 138, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.S.; Sedransk, N.; Volk, H.; Segaloff, A.; Kelley, R.M.; Haines, C.R. Combined androgen and antimetabolite therapy of advanced female breast cancer. A report of the cooperative breast cancer group. Cancer 1975, 36, 308–310. [Google Scholar] [CrossRef]
- Panet-Raymond, V.; Gottlieb, B.; Beitel, L.K.; Pinsky, L.; Trifiro, M.A. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol. Cell. Endocrinol. 2000, 167, 139–150. [Google Scholar] [CrossRef]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Hughes-Davies, L. A functionally significant cross-talk between androgen receptor and ErbB2 pathways in estrogen receptor negative breast cancer. Neoplasia 2008, 10, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; D'Amato, N.C.; Gordon, M.A.; Lind, H.T.; Spoelstra, N.S.; Babbs, B.L.; Heinz, R.E.; Elias, A.; Jedlicka, P.; Jacobsen, B.M.; et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Rondon-Lagos, M.; Villegas, V.E.; Rangel, N.; Sanchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M.; Lombet, A.; Perrot, J.Y.; Jacob, D.; Carvajal, S.; Kazem, A.; Rostene, W.; Therwath, A.; Gompel, A. Proapoptotic effects of antiestrogens, progestins and androgen in breast cancer cells. J. Steroid Biochem. Mol. Biol. 1999, 69, 463–471. [Google Scholar] [CrossRef]
- Lapointe, J.; Fournier, A.; Richard, V.; Labrie, C. Androgens down-regulate bcl-2 protooncogene expression in ZR-75-1 human breast cancer cells. Endocrinology 1999, 140, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Britton, D.J.; Hutcheson, I.R.; Knowlden, J.M.; Barrow, D.; Giles, M.; McClelland, R.A.; Gee, J.M.; Nicholson, R.I. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res. Treat. 2006, 96, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.V.; Leo, M.E.; Tindall, D.J. Modulation of androgen receptor transcriptional activity by the estrogen receptor. J. Androl. 1994, 15, 534–542. [Google Scholar] [PubMed]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Rechoum, Y.; Rovito, D.; Iacopetta, D.; Barone, I.; Ando, S.; Weigel, N.L.; O'Malley, B.W.; Brown, P.H.; Fuqua, S.A. AR collaborates with ERalpha in aromatase inhibitor-resistant breast cancer. Breast Cancer Res. Treat. 2014, 147, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D'Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [PubMed]
- Toth-Fejel, S.; Cheek, J.; Calhoun, K.; Muller, P.; Pommier, R.F. Estrogen and androgen receptors as comediators of breast cancer cell proliferation: Providing a new therapeutic tool. Arch. Surg. 2004, 139, 50–54. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, F.; Thirugnansampanthan, J.; Cui, Y.; Selever, J.; Beyer, A.; Parra, I.; Weigel, N.L.; Herynk, M.H.; Tsimelzon, A.; Lewis, M.T.; et al. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ciupek, A.; Rechoum, Y.; Gu, G.; Gelsomino, L.; Beyer, A.R.; Brusco, L.; Covington, K.R.; Tsimelzon, A.; Fuqua, S.A. Androgen receptor promotes tamoxifen agonist activity by activation of EGFR in ERalpha-positive breast cancer. Breast Cancer Res. Treat. 2015, 154, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Mao, Y.; Fei, X.C.; Shen, K.W. The impact of androgen receptor expression on breast cancer survival: A retrospective study and meta-analysis. PLoS ONE 2013, 8, e82650. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Abduljabbar, R.; Ashankyty, I.; Elmouna, A.; Jerjees, D.; Ali, S.; Buluwela, L.; Diez-Rodriguez, M.; Caldas, C.; Green, A.R.; et al. Prognostic significance of androgen receptor expression in invasive breast cancer: Transcriptomic and protein expression analysis. Breast Cancer Res. Treat. 2016, 159, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wenhui, Z.; Shuo, L.; Dabei, T.; Ying, P.; Zhipeng, W.; Lei, Z.; Xiaohui, H.; Jingshu, G.; Hongtao, S.; Qingyuan, Z. Androgen receptor expression in male breast cancer predicts inferior outcome and poor response to tamoxifen treatment. Eur. J. Endocrinol. 2014, 171, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, E.; Teresiak, M.; Filas, V.; Karczewska, A.; Breborowicz, D.; Mackiewicz, A. BRCA2 mutations and androgen receptor expression as independent predictors of outcome of male breast cancer patients. Clin. Cancer Res. 2003, 9, 4452–4459. [Google Scholar] [PubMed]
- Pich, A.; Margaria, E.; Chiusa, L.; Candelaresi, G.; Dal Canton, O. Androgen receptor expression in male breast carcinoma: Lack of clinicopathological association. Br. J. Cancer. 1999, 79, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Munoz, F.; Quevedo, C.; Martin, M.E.; Alcazar, A.; Salinas, M.; Fando, J.L. Increased activity of eukaryotic initiation factor 2B in PC12 cells in response to differentiation by nerve growth factor. J. Neurochem. 1998, 71, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Castellano, I.; Allia, E.; Accortanzo, V.; Vandone, A.M.; Chiusa, L.; Arisio, R.; Durando, A.; Donadio, M.; Bussolati, G.; Coates, A.S.; et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res. Treat. 2010, 124, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Elebro, K.; Borgquist, S.; Simonsson, M.; Markkula, A.; Jirstrom, K.; Ingvar, C.; Rose, C.; Jernstrom, H. Combined Androgen and Estrogen Receptor Status in Breast Cancer: Treatment Prediction and Prognosis in a Population-Based Prospective Cohort. Clin. Cancer Res. 2015, 21, 3640–3650. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.S.; Kuang, X.Y.; Sun, W.L.; Xu, Y.; Zheng, Y.Z.; Liu, Y.R.; Lang, G.T.; Qiao, F.; Hu, X.; Shao, Z.M. Androgen receptor expression predicts different clinical outcomes for breast cancer patients stratified by hormone receptor status. Oncotarget. 2016, 7, 41285–41293. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.; Tan, L.K.; Chen, B. Correlation of HER-2 status with estrogen and progesterone receptors and histologic features in 3,655 invasive breast carcinomas. Am. J. Clin. Pathol. 2005, 123, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, J.; Koibuchi, Y.; Iijima, K.; Yoshida, T.; Yoshida, M.; Takata, D.; Oyama, T.; Iino, Y.; Morishita, Y. Immunohistochemical double staining with estrogen receptor and HER2 on primary breast cancer. Int. J. Mol. Med. 2003, 12, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Sanga, S.; Broom, B.M.; Cristini, V.; Edgerton, M.E. Gene expression meta-analysis supports existence of molecular apocrine breast cancer with a role for androgen receptor and implies interactions with ErbB family. BMC Med. Genom. 2009. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Vivanco, I.; Kwon, A.; Tran, C.; Wongvipat, J.; Sawyers, C.L. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 2004, 6, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Chia, K.M.; Liu, J.; Francis, G.D.; Naderi, A. A feedback loop between androgen receptor and ERK signaling in estrogen receptor-negative breast cancer. Neoplasia 2011, 13, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, K.; Isotani, S.; Wang, R.; Fujisawa, M.; Gotoh, A.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Soluble factors derived from stroma activated androgen receptor phosphorylation in human prostate LNCaP cells: Roles of ERK/MAP kinase. Prostate 2009, 69, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Mirosevich, J.; Gao, N.; Gupta, A.; Shappell, S.B.; Jove, R.; Matusik, R.J. Expression and role of Foxa proteins in prostate cancer. Prostate 2006, 66, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Chen, Y.; Fei, T.; Li, D.; Lim, E.; Liu, X.S.; Brown, M. Amplitude modulation of androgen signaling by c-MYC. Genes Dev. 2013, 27, 734–748. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Bouchal, J.; Burkadze, G.; Kolar, Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology 2006, 73, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Garay, J.P.; Karakas, B.; Abukhdeir, A.M.; Cosgrove, D.P.; Gustin, J.P.; Higgins, M.J.; Konishi, H.; Konishi, Y.; Lauring, J.; Mohseni, M.; et al. The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Wasielewski, M.; Elstrodt, F.; Klijn, J.G.; Berns, E.M.; Schutte, M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res. Treat. 2006, 99, 97–101. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Chandarlapaty, S.; Ye, Q.; Lobo, J.; Haskell, K.M.; Leander, K.R.; DeFeo-Jones, D.; Huber, H.E.; Rosen, N. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS ONE 2008, 3, e3065. [Google Scholar] [CrossRef] [PubMed]
- Schippinger, W.; Regitnig, P.; Dandachi, N.; Wernecke, K.D.; Bauernhofer, T.; Samonigg, H.; Moinfar, F. Evaluation of the prognostic significance of androgen receptor expression in metastatic breast cancer. Virchows Arch. 2006, 449, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Elsheikh, S.E.; Aleskandarany, M.A.; Habashi, H.O.; Green, A.R.; Powe, D.G.; El-Sayed, M.E.; Benhasouna, A.; Brunet, J.S.; Akslen, L.A.; et al. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Clin. Cancer Res. 2009, 15, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Kassam, F.; Enright, K.; Dent, R.; Dranitsaris, G.; Myers, J.; Flynn, C.; Fralick, M.; Kumar, R.; Clemons, M. Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clin. Breast Cancer 2009, 9, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23 (Suppl. S6), vi7–vi12. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Lara-Medina, F.; Perez-Sanchez, V.; Saavedra-Perez, D.; Blake-Cerda, M.; Arce, C.; Motola-Kuba, D.; Villarreal-Garza, C.; Gonzalez-Angulo, A.M.; Bargallo, E.; Aguilar, J.L.; et al. Triple-negative breast cancer in Hispanic patients: High prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer 2011, 117, 3658–3669. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Kushi, L.H.; Weltzien, E.; Maring, B.; Kutner, S.E.; Fulton, R.S.; Lee, M.M.; Ambrosone, C.B.; Caan, B.J. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Metzger-Filho, O.; Tutt, A.; de Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A., Jr.; Ellis, P.; et al. Dissecting the heterogeneity of triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Dressler, L.G.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Lluch, A.; Albanell, J.; Barry, W.T.; Fan, C.; Chacon, J.I.; Parker, J.S.; Calvo, L.; Plazaola, A.; Arcusa, A.; et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br. J. Cancer. 2014, 111, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J. Clin. Oncol. 2015, 33, 13–21. [Google Scholar] [PubMed]
- Bastien, R.R.; Rodriguez-Lescure, A.; Ebbert, M.T.; Prat, A.; Munarriz, B.; Rowe, L.; Miller, P.; Ruiz-Borrego, M.; Anderson, D.; Lyons, B.; et al. PAM50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med. Genom. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.D.; Zhu, R.; Zhan, M.; Rodriguez, A.A.; Yang, W.; Wong, S.; Makris, A.; Lehmann, B.D.; Chen, X.; Mayer, I.; et al. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 2013, 19, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Jezequel, P.; Loussouarn, D.; Guerin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verriele, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gomez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Hai, E.; Matsumoto, K.; Ikeda, K.; Tokunaga, S.; Nagahara, H.; Sakurai, K.; Inoue, T.; Nishiguchi, Y. Androgen receptor expression in breast cancer: Relationship with clinicopathological factors and biomarkers. Int. J. Clin. Oncol. 2008, 13, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Shi, Y.X.; Li, Z.M.; Jiang, W.Q. Expression and clinical significance of androgen receptor in triple negative breast cancer. Chin. J. Cancer 2010, 29, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sutton, L.M.; Cao, D.; Sarode, V.; Molberg, K.H.; Torgbe, K.; Haley, B.; Peng, Y. Decreased androgen receptor expression is associated with distant metastases in patients with androgen receptor-expressing triple-negative breast carcinoma. Am. J. Clin. Pathol. 2012, 138, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Darb-Esfahani, S.; Denkert, C.; Stenzinger, A.; Salat, C.; Sinn, B.; Schem, C.; Endris, V.; Klare, P.; Schmitt, W.; Blohmer, J.U.; et al. Role of TP53 mutations in triple negative and HER2-positive breast cancer treated with neoadjuvant anthracycline/taxane-based chemotherapy. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Peng, R.; Yuan, Z.; Wang, S.; Peng, J.; Lin, G.; Jiang, X.; Qin, T. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med. Oncol. 2012, 29, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Xu, S.; Zhang, Q.; Zhao, W. The expression and clinical significance of the androgen receptor and E-cadherin in triple-negative breast cancer. Med. Oncol. 2012, 29, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Muller, B.M.; von Minckwitz, G.; Schwabe, M.; Roller, M.; Darb-Esfahani, S.; Ataseven, B.; du Bois, A.; Fissler-Eckhoff, A.; Gerber, B.; et al. Androgen receptor expression in primary breast cancer and its predictive and prognostic value in patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2011, 130, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Pistelli, M.; Caramanti, M.; Biscotti, T.; Santinelli, A.; Pagliacci, A.; De Lisa, M.; Ballatore, Z.; Ridolfi, F.; Maccaroni, E.; Bracci, R.; et al. Androgen receptor expression in early triple-negative breast cancer: Clinical significance and prognostic associations. Cancers 2014, 6, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.S.; Kim, M.S.; Park, H.S.; Lee, J.S.; Lee, J.S.; Kim, S.I.; Park, B.W.; Lee, K.S. Androgen receptor expression is significantly associated with better outcomes in estrogen receptor-positive breast cancers. Ann. Oncol. 2011, 22, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Palla, S.L.; Carey, M.; Agarwal, R.; Meric-Berstam, F.; Traina, T.A.; Hudis, C.; Hortobagyi, G.N.; Gerald, W.L.; et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin. Cancer Res. 2009, 15, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Kashiwagi, S.; Onoda, N.; Kurata, K.; Morisaki, T.; Noda, S.; Takashima, T.; Ohsawa, M.; Kitagawa, S.; Hirakawa, K. Clinical verification of sensitivity to preoperative chemotherapy in cases of androgen receptor-expressing positive breast cancer. Br. J. Cancer 2016, 114, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, W.; Wali, V.B.; Pongor, L.S.; Li, C.; Lau, R.; Gyorffy, B.; Lifton, R.P.; Symmans, W.F.; Pusztai, L.; et al. Predictors of Chemosensitivity in Triple Negative Breast Cancer: An Integrated Genomic Analysis. PLoS Med. 2016, 13, e1002193. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Macarthur, S.; Ross-Innes, C.S.; Tilley, W.D.; Neal, D.E.; Mills, I.G.; Carroll, J.S. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 2011, 30, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Furr, B.J.; Valcaccia, B.; Curry, B.; Woodburn, J.R.; Chesterson, G.; Tucker, H. ICI 176,334: A novel non-steroidal, peripherally selective antiandrogen. J. Endocrinol. 1987, 113, R7–R9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Li, Y.; Song, W.; Xu, Y.; Yang, F.; Zhang, W.; Yin, Y.; Guan, X. Antiproliferative Effect of Androgen Receptor Inhibition in Mesenchymal Stem-Like Triple-Negative Breast Cancer. Cell. Physiol. Biochem. 2016, 38, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Yacoub, R.; Taliaferro-Smith, L.; Osunkoya, A.O.; Odero-Marah, V.A.; Liu, T.; Kimbro, K.S.; Sharma, D.; O’Regan, R.M. Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res. Treat. 2010, 123, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Asthana, S.; Mandal, C.C.; Saxena, S. A molecular analysis provides novel insights into androgen receptor signalling in breast cancer. PLoS ONE 2015, 10, e0120622. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Lopez, M.D.; Montero, J.C.; Morales, J.C.; Prat, A.; Pandiella, A.; Ocana, A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.; Fantl, V.; Gillett, C.; Brookes, S.; Bartek, J.; Smith, R.; Fisher, C.; Barnes, D.; Peters, G. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995, 90, 43–50. [Google Scholar] [CrossRef]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). Palbociclib in Combination with Bicalutamide for the Treatment of AR+ Metastatic Breast Cancer. NCT02605486. Available online: https://clinicaltrials.gov/ct2/show/NCT02605486 (accessed on 9 November 2016).
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Arce-Salinas, C.; Riesco-Martinez, M.C.; Hanna, W.; Bedard, P.; Warner, E. Complete Response of Metastatic Androgen Receptor-Positive Breast Cancer to Bicalutamide: Case Report and Review of the Literature. J. Clin. Oncol. 2016, 34, e21–e24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.P.; He, G.F. A phase II clinical trial of flutamide in the treatment of advanced breast cancer. Tumori 1988, 74, 53–56. [Google Scholar] [PubMed]
- Perrault, D.J.; Logan, D.M.; Stewart, D.J.; Bramwell, V.H.; Paterson, A.H.; Eisenhauer, E.A. Phase II study of flutamide in patients with metastatic breast cancer. A National Cancer Institute of Canada Clinical Trials Group study. Investig. New Drugs 1988, 6, 207–210. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, N.C.; Gordon, M.A.; Babbs, B.; Spoelstra, N.S.; Carson Butterfield, K.T.; Torkko, K.C.; Phan, V.T.; Barton, V.N.; Rogers, T.J.; Sartorius, C.A.; et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol. Cancer Res. 2016, 14, 154–1067. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.J.; Cai, S.; Cichewicz, R.H.; Mooberry, S.L. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res. Treat. 2016, 157, 475–488. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). Taselisib and Enzalutamide in Treating Patients with Androgen Receptor Positive Triple-Negative Metastatic Breast Cancer. NCT 02457910. Available online: https://clinicaltrials.gov/ct2/show/NCT02457910 (accessed on 9 November 2016).
- Ardiani, A.; Farsaci, B.; Rogers, C.J.; Protter, A.; Guo, Z.; King, T.H.; Apelian, D.; Hodge, J.W. Combination therapy with a second-generation androgen receptor antagonist and a metastasis vaccine improves survival in a spontaneous prostate cancer model. Clin. Cancer Res. 2013, 19, 6205–6218. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Ardiani, A.; Gameiro, S.R.; Richards, J.; Hall, A.B.; Hodge, J.W. Androgen deprivation therapy sensitizes triple negative breast cancer cells to immune-mediated lysis through androgen receptor independent modulation of osteoprotegerin. Oncotarget 2016, 7, 23498–23511. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Ardiani, A.; Dirmeier, U.; Wottawah, C.; Schlom, J.; Hodge, J.W. A poxviral-based cancer vaccine the transcription factor twist inhibits primary tumor growth and metastases in a model of metastatic breast cancer and improves survival in a spontaneous prostate cancer model. Oncotarget 2015, 6, 28194–28210. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). A Study to Assess the Efficacy and Safety of Enzalutamide with Trastuzumab in Subjects with Human Epidermal Growth Factor Receptor 2 Positive (HER2+), Androgen Receptor Positive (AR+) Metastatic or Locally Advanced Breast Cancer. NCT02091960. Available online: https://clinicaltrials.gov/ct2/show/NCT02091960 (accessed on 9 November 2016).
- Traina, T.A. Results from a phase 2 study of enzalutamide (Enza), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Parker, J.S.; Peterson, A.C.; Tudor, I.C.; Hoffman, J.; Uppal, H. A novel biomarker to predict sensitivity to enzalutamide in TNBC. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- National Cancer Institute (NCI). Phase IIB Neoadjuvant Enzalutamide (ZT) Plus Taxol for Androgen Receptor (AR)-Positive Triple-Negative Breast Cancer (AR+ TNBC). NCT02689427. Available online: https://clinicaltrials.gov/ct2/show/NCT02689427 (accessed on 9 November 2016).
- National Cancer Institute (NCI). Feasibility Study of Adjuvant Enzalutamide for the Treatment of Early Stage AR (+) Triple Negative Breast Cancer. NCT02750358. Available online: https://clinicaltrials.gov/ct2/show/NCT02750358 (accessed on 9 November 2016).
- Barrie, S.E.; Potter, G.A.; Goddard, P.M.; Haynes, B.P.; Dowsett, M.; Jarman, M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase). J. Steroid Biochem. Mol. Biol. 1994, 50, 267–273. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Campone, M.; Brain, E.; Neven, P.; Hayes, D.; Bondarenko, I.; Griffin, T.W.; Martin, J.; De Porre, P.; Kheoh, T.; et al. Abiraterone acetate, exemestane or the combination in postmenopausal patients with estrogen receptor-positive metastatic breast cancer. Ann. Oncol. 2016, 27, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; O’Shaughnessy, J.A.; Hayes, D.F.; Campone, M.; Bondarenko, I.; Zbarskaya, I.; Brain, E.; Stenina, M.; Ivanova, O.; Graas, M.P.; et al. Biomarker Associations with Efficacy of Abiraterone Acetate and Exemestane in Postmenopausal Patients with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 6002–6009. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). Abiraterone Acetate in Treating Postmenopausal Women with Advanced or Metastatic breast Cancer. NCT00755885. Available online: https://clinicaltrials.gov/ct2/show/NCT00755885 (accessed on 9 November 2016).
- National Cancer Institute (NCI). AZD8186 First Time in Patient Ascending Dose Study. NCT01884285. Available online: https://clinicaltrials.gov/ct2/show/NCT01884285 (accessed on 9 November 2016).
- Fizazi, K.; Jones, R.; Oudard, S.; Efstathiou, E.; Saad, F.; de Wit, R.; De Bono, J.; Cruz, F.M.; Fountzilas, G.; Ulys, A.; et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy: ELM-PC 5. J. Clin. Oncol. 2015, 33, 723–731. [Google Scholar] [PubMed]
- Saad, F.; Fizazi, K.; Jinga, V.; Efstathiou, E.; Fong, P.C.; Hart, L.L.; Jones, R.; McDermott, R.; Wirth, M.; Suzuki, K.; et al. Orteronel plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (ELM-PC 4): A double-blind, multicentre, phase 3, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 338–348. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). Orteronel as Monotherapy in Patients with Metastatic Breast Cancer (MBC) that Expresses the Androgen Receptor (AR). NCT01990209. Available online: https://clinicaltrials.gov/ct2/show/NCT01990209 (accessed on 9 November 2016).
- National Cancer Institute (NCI). A Open-Label Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of VT-464 in Patients with Advanced Breast Cancer. NCT02580448. Available online: https://clinicaltrials.gov/ct2/show/NCT02580448 (accessed on 9 November 2016).
- Kandil, S.; Westwell, A.D.; McGuigan, C. 7-Substituted umbelliferone derivatives as androgen receptor antagonists for the potential treatment of prostate and breast cancer. Bioorg. Med. Chem. Lett. 2016, 26, 2000–2004. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Ahn, S.; Cheney, M.D.; Yepuru, M.; Miller, D.D.; Steiner, M.S.; Dalton, J.T. Selective androgen receptor modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial:mesenchymal stem cell signaling. PLoS ONE 2014, 9, e103202. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). Efficacy and Safety of GTx-024 in Patients with Androgen Receptor-Positive Triple Negative Breast Cancer (AR+ TNBC). NCT02368691. Available online: https://clinicaltrials.gov/ct2/show/NCT02368691 (accessed on 9 November 2016).
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Irvine, R.A.; Buchanan, G.; Koh, S.S.; Park, J.M.; Tilley, W.D.; Stallcup, M.R.; Press, M.F.; Coetzee, G.A. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res. 2000, 60, 5946–5949. [Google Scholar] [PubMed]
- Shin, S.; Verma, I.M. BRCA2 cooperates with histone acetyltransferases in androgen receptor-mediated transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 7201–7206. [Google Scholar] [CrossRef] [PubMed]
- Berns, E.M.; Dirkzwager-Kiel, M.J.; Kuenen-Boumeester, V.; Timmermans, M.; Verhoog, L.C.; van den Ouweland, A.M.; Meijer-Heijboer, H.; Klijn, J.G.; van der Kwast, T.H. Androgen pathway dysregulation in BRCA1-mutated breast tumors. Breast Cancer Res. Treat. 2003, 79, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Pristauz, G.; Petru, E.; Stacher, E.; Geigl, J.B.; Schwarzbraun, T.; Tsybrovskyy, O.; Winter, R.; Moinfar, F. Androgen receptor expression in breast cancer patients tested for BRCA1 and BRCA2 mutations. Histopathology 2010, 57, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- McCune, K.; Bhat-Nakshatri, P.; Thorat, M.A.; Nephew, K.P.; Badve, S.; Nakshatri, H. Prognosis of hormone-dependent breast cancers: Implications of the presence of dysfunctional transcriptional networks activated by insulin via the immune transcription factor T-bet. Cancer Res. 2010, 70, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Biarnes, M.C.; Clarke, R.; Jordan, V.C. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res. Treat. 2015, 150, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Bihani, T.; Ezell, S.A.; Ladd, B.; Grosskurth, S.E.; Mazzola, A.M.; Pietras, M.; Reimer, C.; Zinda, M.; Fawell, S.; D’Cruz, C.M. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget 2015, 6, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Borbely, G.; Haldosen, L.A.; Dahlman-Wright, K.; Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015, 6, 33623–33635. [Google Scholar] [PubMed]
- Perez-Pena, J.; Serrano-Heras, G.; Montero, J.C.; Corrales-Sanchez, V.; Pandiella, A.; Ocana, A. In Silico Analysis Guides Selection of BET Inhibitors for Triple-Negative Breast Cancer Treatment. Mol. Cancer Ther. 2016, 15, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Sahni, J.M.; Gayle, S.S.; Bonk, K.L.; Vite, L.C.; Yori, J.L.; Webb, B.; Ramos, E.K.; Seachrist, D.D.; Landis, M.D.; Chang, J.C.; et al. Bromodomain and Extraterminal Protein Inhibition Blocks Growth of Triple-negative Breast Cancers through the Suppression of Aurora Kinases. J. Biol. Chem. 2016, 291, 23756–23768. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). A Dose-Finding Study of OTX105/MK-8628, a Small Molecule Inhibitor of the Bromodomain and Extra-Terminal (BET) Proteins, in Adults with Selected Advanced Solid Tumors (MK-8628-003). NCT02259114. Available online: https://clinicaltrials.gov/ct2/show/NCT02259114 (accessed on 9 November 2016).
- National Cancer Institute (NCI). A Dose Exploration Study with MK-8628 in Participants with Selected Advanced Solid Tumors (MK-8628-006). NCT02698176. Available online: https://clinicaltrials.gov/ct2/show/NCT02698176 (accessed on 9 November 2016).
- National Cancer Institute (NCI). A Study to Investigate Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Activity of GSK525762 in Subjects with NUT Midline Carcinoma (NMC) and Other Cancers. NCT01587703. Available online: https://clinicaltrials.gov/ct2/show/NCT01587703 (accessed on 9 November 2016).
- Burdall, S.E.; Hanby, A.M.; Lansdown, M.R.; Speirs, V. Breast cancer cell lines: Friend or foe? Breast Cancer Res. 2003, 5, 89–95. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trial ID | Agent(s) | Mechanism(s) of Action | Patient Population | Study Design |
|---|---|---|---|---|
| NCT02605486 | Palbociclib & Bicalutamide | CD4/CD6 Inhibitor & Androgen Receptor Inhibitor | AR-positive metastatic breast cancer | Non-randomized, open-label, phase I/II |
| NCT02457910 | Taselisib & Enzalutamide | PI3K Inhibitor & Androgen Receptor Inhibitor | AR-positive metastatic TNBC | Partially-randomized, open-label phase IB/II |
| NCT02091960 | Enzalutamide & Trastuzumab | Androgen Receptor Inhibitor & HER2 Targeted Monoclonal Antibody | AR-positive, HER2 amplified metastatic or locally advanced breast cancer | Non-randomized, open label, phase II |
| NCT02689427 | Enzalutamide & Paclitaxel | Androgen Receptor Inhibitor & Microtubule Stabilizer | AR-positive TNBC, stage I–III breast cancer (neoadjuvant therapy) | Non-randomized, open label, phase IIB |
| NCT02750358 | Enzalutamide | Androgen Receptor Inhibitor | AR-positive TNBC, stage I–III breast cancer (adjuvant therapy) | Non-randomized, open-label, feasibility study |
| NCT00755885 | Abiraterone Acetate | CYP17 Inhibitor | ER or AR-positive postmenopausal metastatic or locally advanced breast cancer | Non-randomized, open-label, phase I/II |
| NCT01884285 | AZD8186 +/− Abiraterone Acetate or AZD2014 | PI3K Inhibitor +/− CYP17 Inhibitor or mTOR Inhibitor | Advanced TNBC | Non-randomized, open-label, phase I |
| NCT01990209 | Orteronel | CYP17 Inhibitor | AR-positive metastatic breast cancer | Non-randomized, open-label, phase II |
| NCT02580448 | VT-464 | CYP17 Inhibitor | Advanced breast cancer. Phase I: TNBC or ER-positive, HER2 negative | Non-randomized, open-label, phase I/II |
| Phase II: AR-positive TNBC or ER-positive, HER2 negative | ||||
| NCT02368691 | GTx-024 | Selective Androgen Receptor Modulator | AR-positive advanced TNBC | Non-randomized, open-label, phase II |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahim, B.; O’Regan, R. AR Signaling in Breast Cancer. Cancers 2017, 9, 21. https://doi.org/10.3390/cancers9030021
Rahim B, O’Regan R. AR Signaling in Breast Cancer. Cancers. 2017; 9(3):21. https://doi.org/10.3390/cancers9030021
Chicago/Turabian StyleRahim, Bilal, and Ruth O’Regan. 2017. "AR Signaling in Breast Cancer" Cancers 9, no. 3: 21. https://doi.org/10.3390/cancers9030021
APA StyleRahim, B., & O’Regan, R. (2017). AR Signaling in Breast Cancer. Cancers, 9(3), 21. https://doi.org/10.3390/cancers9030021