
Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer


Abstract
:1. Introduction
2. AR Structure and Collaborating Factors in AR Signaling Pathway
3. The Unique Features of Transcription Factors in Castration-Resistant Prostate Cancer
4. Development of Novel Drugs
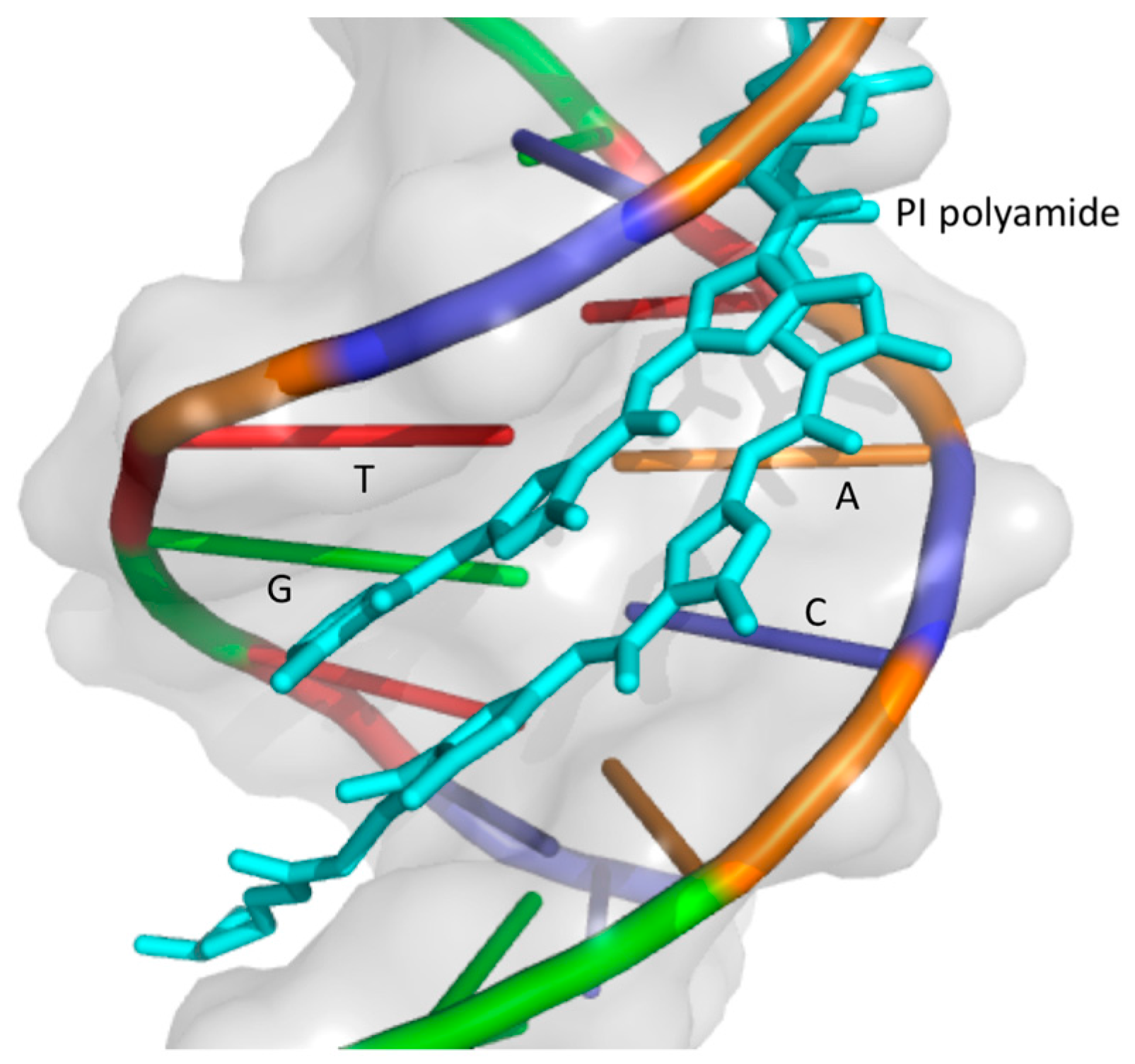
4.1. Pyrrole-Imidazole Polyamide
4.2. Novel Drugs Targeting TFs Related to the AR Pathway
4.2.1. The Pioneer Factors (FOXA1 and GATA2)
4.2.2. OCT1
4.2.3. ETS Family Genes
4.2.4. NKX3-1
4.2.5. C/EBP Family
4.2.6. E2F-1
4.2.7. c-MYC
4.2.8. STAT3
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hobisch, A.; Culig, Z.; Radmayr, C.; Bartsch, G.; Klocker, H.; Hittmair, A. Distant metastases from prostatic carcinoma express androgen receptor protein. Cancer Res. 1995, 55, 3068–3072. [Google Scholar] [PubMed]
- Hobisch, A.; Culig, Z.; Radmayr, C.; Bartsch, G.; Klocker, H.; Hittmair, A. Androgen receptor status of lymph node metastases from prostate cancer. Prostate 1996, 28, 129–135. [Google Scholar] [CrossRef]
- Sadi, M.V.; Walsh, P.C.; Barrack, E.R. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer 1991, 67, 3057–3064. [Google Scholar] [CrossRef]
- Tilley, W.D.; Lim-Tio, S.S.; Horsfall, D.J.; Aspinall, J.O.; Marshall, V.R.; Skinner, J.M. Detection of discrete androgen receptor epitopes in prostate cancer by immunostaining: Measurement by color video image analysis. Cancer Res. 1994, 54, 4096–4102. [Google Scholar] [PubMed]
- Van der Kwast, T.H.; Tetu, B. Androgen receptors in untreated and treated prostatic intraepithelial neoplasia. Eur. Urol. 1996, 30, 265–268. [Google Scholar] [PubMed]
- Huggins, C. Effect of Orchiectomy and Irradiation on Cancer of the Prostate. Ann. Surg. 1942, 115, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Trapman, J.; Brinkmann, A.O. The androgen receptor in prostate cancer. Pathol. Res. Pract. 1996, 192, 752–760. [Google Scholar] [CrossRef]
- Taplin, M.E.; Balk, S.P. Androgen receptor: A key molecule in the progression of prostate cancer to hormone independence. J. Cell. Biochem. 2004, 91, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.T.; Isaacs, J.T. Mechanisms involved in the progression of androgen-independent prostate cancers: It is not only the cancer cell’s fault. Endocr. Relat. Cancer 2002, 9, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Hoffmann, J.; Erdel, M.; Eder, I.E.; Hobisch, A.; Hittmair, A.; Bartsch, G.; Utermann, G.; Schneider, M.R.; Parczyk, K.; et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 1999, 81, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xie, C.C.; Zhu, Y.; Li, T.; Sun, J.; Cheng, Y.; Ewing, C.M.; Dalrymple, S.; Turner, A.R.; Sun, J.; et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia 2008, 10, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Regan, K.M.; Schmidt, L.J.; Anderson, S.K.; Ballman, K.V.; Tindall, D.J. Androgen modulation of coregulator expression in prostate cancer cells. Mol. Endocrinol. 2009, 23, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Ford, O.H., 3rd; Gregory, C.W.; Kim, D.; Smitherman, A.B.; Mohler, J.L. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J. Urol. 2003, 170, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, M.; Hara, T.; Kusaka, M. Overcoming persistent dependency on androgen signaling after progression to castration-resistant prostate cancer. Clin. Cancer Res. 2010, 16, 4319–4324. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Antonarakis, E.S. Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: A new era of hormonal therapies is born. Ther. Adv. Urol. 2012, 4, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; O’Malley, B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G.; van der Korput, H.A.; Trapman, J.; Brinkmann, A.O. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem. 1995, 270, 7341–7346. [Google Scholar] [PubMed]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp, M.P.; O'Mahony, O.A.; Brogley, M.; Rehman, H.; Lapensee, E.W.; Dhanasekaran, S.; Hofer, M.D.; Kuefer, R.; Chinnaiyan, A.; Rubin, M.A.; et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009, 69, 4434–4442. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.; Dong, Y.; Zhang, H.; Qi, Y.; Balk, S.P.; Sartor, O. Castration-resistant prostate cancer: Adaptive responses in the androgen axis. Cancer Treat. Rev. 2014, 40, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Schrecengost, R.; Knudsen, K.E. Molecular pathogenesis and progression of prostate cancer. Semin. Oncol. 2013, 40, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Middleman, M.N.; Lush, R.M.; Figg, W.D. The mutated androgen receptor and its implications for the treatment of metastatic carcinoma of the prostate. Pharmacotherapy 1996, 16, 376–381. [Google Scholar] [PubMed]
- Knudsen, K.E.; Penning, T.M. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, J.P.; McLeod, D.G.; Heidenberg, H.B.; Sesterhenn, I.A.; Finger, M.J.; Moul, J.W.; Srivastava, S. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res. 1994, 54, 2861–2864. [Google Scholar] [PubMed]
- Taplin, M.E.; Bubley, G.J.; Ko, Y.J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999, 59, 2511–2515. [Google Scholar] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Wu, J.H.; Trifiro, M. The androgen receptor gene mutations database (ARDB): 2004 update. Hum. Mutat. 2004, 23, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar] [PubMed]
- Takayama, K.; Horie-Inoue, K.; Katayama, S.; Suzuki, T.; Tsutsumi, S.; Ikeda, K.; Urano, T.; Fujimura, T.; Takagi, K.; Takahashi, S.; et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 2013, 32, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Suzuki, T.; Fujimura, T.; Urano, T.; Takahashi, S.; Homma, Y.; Inoue, S. CtBP2 modulates the androgen receptor to promote prostate cancer progression. Cancer Res. 2014, 74, 6452–6453. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Structure of chromatin. Annu. Rev. Biochem. 1977, 46, 931–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Janne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed]
- Bossard, P.; Zaret, K.S. GATA transcription factors as potentiators of gut endoderm differentiation. Development 1998, 125, 4909–4917. [Google Scholar] [PubMed]
- Cuesta, I.; Zaret, K.S.; Santisteban, P. The forkhead factor FoxE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Mol. Cell. Biol. 2007, 27, 7302–7314. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; Zaret, K.S. Specific interactions of the wing domains of FOXA1 transcription factor with DNA. J. Mol. Biol. 2007, 366, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Zhang, J.; Rao, M.A.; Case, T.C.; Mirosevich, J.; Wang, Y.; Jin, R.; Gupta, A.; Rennie, P.S.; Matusik, R.J. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003, 17, 1484–1507. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Muntener, M.; Kristiansen, G. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Turbin, D.; Thorat, M.A.; Morimiya, A.; Nielsen, T.O.; Perou, C.M.; Dunn, S.; Huntsman, D.G.; Nakshatri, H. FOXA1 expression in breast cancer—Correlation with luminal subtype A and survival. Clin. Cancer Res. 2007, 13, 4415–4421. [Google Scholar] [CrossRef] [PubMed]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoute, J.; Keeton, E.K.; Lupien, M.; Krum, S.A.; Carroll, J.S.; Brown, M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007, 67, 6477–6483. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Locke, W.J.; Sutherland, R.L.; Kench, J.G.; Henshall, S.M. A role for GATA-2 in transition to an aggressive phenotype in prostate cancer through modulation of key androgen-regulated genes. Oncogene 2009, 28, 3847–3856. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sunkel, B.; Chen, Z.; Liu, X.; Ye, Z.; Li, Q.; Grenade, C.; Ke, J.; Zhang, C.; Chen, H.; et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014, 42, 3607–3622. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, C.; Wu, D.; Chen, H.; Rorick, A.; Zhang, X.; Wang, Q. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J. 2011, 30, 2405–2419. [Google Scholar] [CrossRef] [PubMed]
- Hagege, H.; Klous, P.; Braem, C.; Splinter, E.; Dekker, J.; Cathala, G.; de Laat, W.; Forne, T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2007, 2, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Klemm, J.D.; Rould, M.A.; Aurora, R.; Herr, W.; Pabo, C.O. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell 1994, 77, 21–32. [Google Scholar] [CrossRef]
- Jariwala, U.; Cogan, J.P.; Jia, L.; Frenkel, B.; Coetzee, G.A. Inhibition of AR-mediated transcription by binding of Oct1 to a motif enriched in AR-occupied regions. Prostate 2009, 69, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Berman, B.P.; Jariwala, U.; Yan, X.; Cogan, J.P.; Walters, A.; Chen, T.; Buchanan, G.; Frenkel, B.; Coetzee, G.A. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE 2008, 3, e3645. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Gemberling, M.; Nakamura, M.; Whitby, F.G.; Handa, H.; Fairbrother, W.G.; Tantin, D. A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev. 2009, 23, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Tantin, D.; Schild-Poulter, C.; Wang, V.; Hache, R.J.; Sharp, P.A. The octamer binding transcription factor Oct-1 is a stress sensor. Cancer Res. 2005, 65, 10750–10758. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Sakamoto, S.; Song, D.; Qu, Z.; Ota, K.; Taniguchi, T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998, 424, 27–32. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Takayama, K.; Urano, T.; Murata, T.; Kumagai, J.; Fujimura, T.; Ikeda, K.; Horie-Inoue, K.; Homma, Y.; Ouchi, Y.; et al. Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int. J. Cancer 2012, 130, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Minekura, H.; Kang, M.J.; Inagaki, Y.; Suzuki, H.; Sato, H.; Fujino, T.; Yamamoto, T.T. Genomic organization and transcription units of the human acyl-CoA synthetase 3 gene. Gene 2001, 278, 185–192. [Google Scholar] [CrossRef]
- Obinata, D.; Takayama, K.; Fujiwara, K.; Suzuki, T.; Tsutsumi, S.; Fukuda, N.; Nagase, H.; Fujimura, T.; Urano, T.; Homma, Y.; et al. Targeting Oct1 genomic function inhibits androgen receptor signaling and castration-resistant prostate cancer growth. Oncogene 2016, 35, 6350–6358. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Adryan, B.; Barbosa-Morais, N.L.; Lynch, A.G.; Tran, M.G.; Neal, D.E.; Mills, I.G. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007, 8, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Chen, Y.B.; Banerjee, S.; Pan, Y.; Yu, J.; Vuong, T.; Perner, S.; Lafargue, C.J.; Mertz, K.D.; Setlur, S.R.; et al. ERG cooperates with androgen receptor in regulating trefoil factor 3 in prostate cancer disease progression. Neoplasia 2010, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.Y.; Chang, C.W.; Chng, K.R.; Wansa, K.D.; Sung, W.K.; Cheung, E. Integration of regulatory networks by NKX3–1 promotes androgen-dependent prostate cancer survival. Mol. Cell. Biol. 2012, 32, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; Gong, Y.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Suzuki, T.; Tsutsumi, S.; Fujimura, T.; Urano, T.; Takahashi, S.; Homma, Y.; Aburatani, H.; Inoue, S. RUNX1, an androgen- and EZH2-regulated gene, has differential roles in AR-dependent and -independent prostate cancer. Oncotarget 2015, 6, 2263–2276. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Suzuki, T.; Tsutsumi, S.; Fujimura, T.; Takahashi, S.; Homma, Y.; Urano, T.; Aburatani, H.; Inoue, S. Integrative analysis of FOXP1 function reveals a tumor-suppressive effect in prostate cancer. Mol. Endocrinol. 2014, 28, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Elliott, A.D.; DeGraff, D.J.; Anderson, P.D.; Anumanthan, G.; Yamashita, H.; Sun, Q.; Friedman, D.B.; Hachey, D.L.; Yu, X.; et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol. Endocrinol. 2014, 28, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gonit, M.; Salazar, M.D.; Shatnawi, A.; Shemshedini, L.; Trumbly, R.; Ratnam, M. C/EBPα redirects androgen receptor signaling through a unique bimodal interaction. Oncogene 2010, 29, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.P.; Watson, D.K. ETS transcription factors: Oncogenes and tumor suppressor genes as therapeutic targets for prostate cancer. Expert Rev. Anticancer Ther. 2008, 8, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, M.L.; Wilson, L.A.; Verschoor, C.P.; Singh, G. Ets-1 regulates energy metabolism in cancer cells. PLoS ONE 2010, 5, e13565. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Findlay, V.J.; Bandurraga, S.G.; Kistner-Griffin, E.; Spruill, L.S.; Liu, A.; Golshayan, A.R.; Turner, D.P. ETS1 transcriptional activity is increased in advanced prostate cancer and promotes the castrate-resistant phenotype. Carcinogenesis 2012, 33, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Preece, D.M.; Harvey, J.M.; Bentel, J.M.; Thomas, M.A. ETS1 regulates NKX3.1 5′ promoter activity and expression in prostate cancer cells. Prostate 2011, 71, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Wotton, K.R.; Weierud, F.K.; Juarez-Morales, J.L.; Alvares, L.E.; Dietrich, S.; Lewis, K.E. Conservation of gene linkage in dispersed vertebrate NK homeobox clusters. Dev. Genes Evol. 2009, 219, 481–496. [Google Scholar] [CrossRef] [PubMed]
- He, W.W.; Sciavolino, P.J.; Wing, J.; Augustus, M.; Hudson, P.; Meissner, P.S.; Curtis, R.T.; Shell, B.K.; Bostwick, D.G.; Tindall, D.J.; et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics 1997, 43, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Muders, M.H.; Li, J.; Rinaldo, F.; Tindall, D.J.; Datta, K. Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer. Cancer Res. 2008, 68, 8770–8778. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Hermans, K.G.; van Marion, R.; van Dekken, H.; Jenster, G.; van Weerden, W.M.; Trapman, J. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006, 66, 10658–10663. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Zheng, T.; Gelmann, E.P. NKX3.1 Suppresses TMPRSS2-ERG Gene Rearrangement and Mediates Repair of Androgen Receptor-Induced DNA Damage. Cancer Res. 2015, 75, 2686–2698. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wang, H.; Xu, Y.; Chen, S.; Balk, S.P. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009, 69, 6027–6032. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tu, S.W.; Hsieh, J.T. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J. Biol. Chem. 2005, 280, 22437–22444. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Ye, H.; He, H.H.; Gerrin, S.J.; Chen, S.; Tanenbaum, B.A.; Cai, C.; Sowalsky, A.G.; He, L.; Wang, H.; et al. SOX9 drives WNT pathway activation in prostate cancer. J. Clin. Investig. 2016, 126, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Kokontis, J.M.; Hay, N.; Liao, S. Progression of LNCaP prostate tumor cells during androgen deprivation: Hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol. 1998, 12, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Huang, D.; Kelly, W.K.; Slovin, S.F.; Stephenson, R.D.; Eicher, C.; Delacruz, A.; Curley, T.; Schwartz, L.H.; Scher, H.I. Phase 1 trial of high-dose exogenous testosterone in patients with castration-resistant metastatic prostate cancer. Eur. Urol. 2009, 56, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell. Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Pourtier-Manzanedo, A.; Gil, J.; Beach, D.H. Myc confers androgen-independent prostate cancer cell growth. J. Clin. Investig. 2003, 112, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Kaltz-Wittmer, C.; Klenk, U.; Glaessgen, A.; Aust, D.E.; Diebold, J.; Lohrs, U.; Baretton, G.B. FISH analysis of gene aberrations (MYC, CCND1, ERBB2, RB, and AR) in advanced prostatic carcinomas before and after androgen deprivation therapy. Lab. Investig. 2000, 80, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Nupponen, N.N.; Kakkola, L.; Koivisto, P.; Visakorpi, T. Genetic alterations in hormone-refractory recurrent prostate carcinomas. Am. J. Pathol. 1998, 153, 141–148. [Google Scholar] [CrossRef]
- Eagle, L.R.; Yin, X.; Brothman, A.R.; Williams, B.J.; Atkin, N.B.; Prochownik, E.V. Mutation of the MXI1 gene in prostate cancer. Nat. Genet. 1995, 9, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ge, K.; Minhas, F.; Duhadaway, J.; Mao, N.C.; Wilson, D.; Buccafusca, R.; Sakamuro, D.; Nelson, P.; Malkowicz, S.B.; Tomaszewski, J.; et al. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int. J. Cancer 2000, 86, 155–161. [Google Scholar] [CrossRef]
- Sun, C.; Dobi, A.; Mohamed, A.; Li, H.; Thangapazham, R.L.; Furusato, B.; Shaheduzzaman, S.; Tan, S.H.; Vaidyanathan, G.; Whitman, E.; et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008, 27, 5348–5353. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK–STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of STAT3 in human prostate tumors and cell lines: Direct inhibition of STAT3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar] [PubMed]
- Culig, Z.; Steiner, H.; Bartsch, G.; Hobisch, A. Interleukin-6 regulation of prostate cancer cell growth. J. Cell. Biochem. 2005, 95, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marie, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6, 7736. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Lu, Y.; Chao, H.P.; Zhang, D.; Liu, X.; Chen, X.; Li, Q.; Rycaj, K.; Calhoun-Davis, T.; et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed]
- Trauger, J.W.; Baird, E.E.; Dervan, P.B. Recognition of DNA by designed ligands at subnanomolar concentrations. Nature 1996, 382, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L.; Bremer, R.E.; White, S.; Szewczyk, J.W.; Turner, J.M.; Baird, E.E.; Dervan, P.B.; Rees, D.C. Structural effects of DNA sequence on T·A recognition by hydroxypyrrole/pyrrole pairs in the minor groove. J. Mol. Biol. 2000, 295, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.B.; Edelson, B.S. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol. 2003, 13, 284–299. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Baird, E.E.; Dervan, P.B.; Rees, D.C. Structural basis for G.C recognition in the DNA minor groove. Nat. Struct. Biol. 1998, 5, 104–109. [Google Scholar] [CrossRef] [PubMed]
- White, S.; Szewczyk, J.W.; Turner, J.M.; Baird, E.E.; Dervan, P.B. Recognition of the four Watson–Crick base pairs in the DNA minor groove by synthetic ligands. Nature 1998, 391, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.L.; Montgomery, D.C.; Dervan, P.B. Enhancing the cellular uptake of Py-Im polyamides through next-generation aryl turns. Nucleic Acids Res. 2012, 40, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bando, T.; Sugiyama, H. Discrimination of hairpin polyamides with an alpha-substituted-gamma-aminobutyric acid as a 5′-TG-3′ reader in DNA minor groove. J. Am. Chem. Soc. 2006, 128, 8766–8776. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, D.M.; Dervan, P.B. Allosteric modulation of DNA by small molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 13175–13179. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L.; White, S.; Szewczyk, J.W.; Turner, J.M.; Baird, E.E.; Dervan, P.B.; Rees, D.C. A structural basis for recognition of A·T and T·A base pairs in the minor groove of B-DNA. Science 1998, 282, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.J.; Cronin, M.T. A review of the electrophilic reaction chemistry involved in covalent DNA binding. Crit. Rev. Toxicol. 2010, 40, 728–748. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Aoyama, T.; Yokoe, T.; Fukasawa, A.; Fukuda, N.; Ueno, T.; Sugiyama, H.; Nagase, H.; Matsumoto, Y. Pharmacokinetic modeling and prediction of plasma pyrrole-imidazole polyamide concentration in rats using simultaneous urinary and biliary excretion data. Biol. Pharm. Bull. 2009, 32, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, A.; Aoyama, T.; Nagashima, T.; Fukuda, N.; Ueno, T.; Sugiyama, H.; Nagase, H.; Matsumoto, Y. Pharmacokinetics of pyrrole-imidazole polyamides after intravenous administration in rat. Biopharm. Drug Dispos. 2009, 30, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Fukuda, N.; Ueno, T.; Tahira, Y.; Ayame, H.; Zhang, W.; Bando, T.; Sugiyama, H.; Saito, S.; Matsumoto, K.; et al. Development of gene silencing pyrrole-imidazole polyamide targeting the TGF-β1 promoter for treatment of progressive renal diseases. J. Am. Soc. Nephrol. 2006, 17, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Fukuda, N.; Ueno, T.; Katakawa, M.; Wang, X.; Watanabe, T.; Matsui, S.; Aoyama, T.; Saito, K.; Bando, T.; et al. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole-imidazole polyamide targeting of the transforming growth factor-β1 promoter. Kidney Int. 2011, 79, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Fukuda, N.; Inoue, T.; Nakai, S.; Saito, K.; Fujiwara, K.; Matsuda, H.; Ueno, T.; Matsumoto, Y.; Watanabe, T.; et al. Preclinical Study of Novel Gene Silencer Pyrrole-Imidazole Polyamide Targeting Human TGF-β1 Promoter for Hypertrophic Scars in a Common Marmoset Primate Model. PLoS ONE 2015, 10, e0125295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, C.; Mitsiades, N. Moving Beyond the Androgen Receptor (AR): Targeting AR-Interacting Proteins to Treat Prostate Cancer. Horm. Cancer 2016, 7, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Zhao, J.C.; Wu, L.; Kim, J.; Yu, J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014, 5, 3972. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Horie-Inoue, K.; Ikeda, K.; Urano, T.; Murakami, K.; Hayashizaki, Y.; Ouchi, Y.; Inoue, S. FOXP1 is an androgen-responsive transcription factor that negatively regulates androgen receptor signaling in prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Fujino, K.; Monteiro, L.J.; Gomes, A.R.; Drost, R.; Davidson-Smith, H.; Takeda, S.; Khoo, U.S.; Jonkers, J.; Sproul, D.; et al. FOXA1 repression is associated with loss of BRCA1 and increased promoter methylation and chromatin silencing in breast cancer. Oncogene 2015, 34, 5012–5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.C.; Fong, K.W.; Jin, H.J.; Yang, Y.A.; Kim, J.; Yu, J. FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene 2016, 35, 4335–4344. [Google Scholar] [CrossRef] [PubMed]
- Umetani, M.; Nakao, H.; Doi, T.; Iwasaki, A.; Ohtaka, M.; Nagoya, T.; Mataki, C.; Hamakubo, T.; Kodama, T. A novel cell adhesion inhibitor, K-7174, reduces the endothelial VCAM-1 induction by inflammatory cytokines, acting through the regulation of GATA. Biochem. Biophys. Res. Commun. 2000, 272, 370–374. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Lanz, R.B.; Fiskus, W.; Geng, C.; Yi, P.; Hartig, S.M.; Rajapakshe, K.; Shou, J.; Wei, L.; Shah, S.S.; et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc. Natl. Acad. Sci. USA 2014, 111, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, S.; Nakano, Y.; Obara, N.; Suzuki, N.; Doi, T.; Kodama, T.; Nagasawa, T.; Yamamoto, M. A GATA-specific inhibitor (K-7174) rescues anemia induced by IL-1β, TNF-α, or L-NMMA. FASEB J. 2003, 17, 1742–1744. [Google Scholar] [PubMed]
- Kikuchi, J.; Yamada, S.; Koyama, D.; Wada, T.; Nobuyoshi, M.; Izumi, T.; Akutsu, M.; Kano, Y.; Furukawa, Y. The novel orally active proteasome inhibitor K-7174 exerts anti-myeloma activity in vitro and in vivo by down-regulating the expression of class I histone deacetylases. J. Biol. Chem. 2013, 288, 25593–25602. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hiramatsu, N.; Okamura, M.; Hayakawa, K.; Shimada, T.; Kasai, A.; Yokouchi, M.; Shitamura, A.; Yao, J.; Paton, A.W.; et al. Suppression of cytokine response by GATA inhibitor K-7174 via unfolded protein response. Biochem. Biophys. Res. Commun. 2007, 360, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. USA 2007, 104, 10418–10423. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Fujiwara, Y.; Doki, Y.; Sugita, Y.; Sohma, I.; Miyata, H.; Takiguchi, S.; Yasuda, T.; Tomita, N.; Morishita, R.; et al. Gene therapy using ETS-1 transcription factor decoy for peritoneal dissemination of gastric cancer. Int. J. Cancer 2007, 121, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J. Transcription factor decoys: A new model for disease intervention. Ann. N. Y. Acad. Sci. 2005, 1058, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Uren, A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Ito, A.; Fujiwara, K.; Takayama, K.; Ashikari, D.; Murata, Y.; Yamaguchi, K.; Urano, T.; Fujimura, T.; Fukuda, N.; et al. Pyrrole-imidazole polyamide targeted to break fusion sites in TMPRSS2 and ERG gene fusion represses prostate tumor growth. Cancer Sci. 2014, 105, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, A.E.; Martinez, T.F.; Hare, A.A.; Kurmis, A.A.; Phillips, J.W.; Sud, S.; Pienta, K.J.; Dervan, P.B. Tumor Repression of VCaP Xenografts by a Pyrrole-Imidazole Polyamide. PLoS ONE 2015, 10, e0143161. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Kang, M.R.; Wang, J.; Huang, V.; Place, R.F.; Sun, Y.; Li, L.C. Targeted induction of endogenous NKX3-1 by small activating RNA inhibits prostate tumor growth. Prostate 2013, 73, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Mintz, P.; Tien, Y.W.; Lai, H.S.; Saetrom, P.; Reccia, I.; Swiderski, P.; Armstrong, B.; et al. Targeted Delivery of C/EBPα -saRNA by Pancreatic Ductal Adenocarcinoma-specific RNA Aptamers Inhibits Tumor Growth In Vivo. Mol. Ther. 2016, 24, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, A.O.; Chinnakannu, K.; Chen, D.; Sivanandam, A.; Tejwani, S.; Menon, M.; Dou, Q.P.; Reddy, G.P. Androgen receptor and E2F-1 targeted thymoquinone therapy for hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7782–7788. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Kerrigan, J.E.; Minko, T.; Garbuzenko, O.; Lee, K.C.; Scarborough, A.; Abali, E.E.; Budak-Alpdogan, T.; Johnson-Farley, N.; Banerjee, D.; et al. Antitumor and modeling studies of a penetratin-peptide that targets E2F-1 in small cell lung cancer. Cancer Biol. Ther. 2013, 14, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Bansal, N.; Shaik, T.; Kerrigan, J.E.; Minko, T.; Garbuzenko, O.; Abali, E.E.; Johnson-Farley, N.; Banerjee, D.; Scotto, K.W.; et al. A novel peptide that inhibits E2F transcription and regresses prostate tumor xenografts. Oncotarget 2014, 5, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, C.; D'Agnano, I.; Lozupone, F.; Valentini, A.; Geiser, T.; Zon, G.; Calabretta, B.; Citro, G.C.; Zupi, G. Antitumor effect of c-myc antisense phosphorothioate oligodeoxynucleotides on human melanoma cells in vitro and and in mice. J. Natl. Cancer Inst. 1996, 88, 419–429. [Google Scholar] [CrossRef] [PubMed]
- McGuffie, E.M.; Catapano, C.V. Design of a novel triple helix-forming oligodeoxyribonucleotide directed to the major promoter of the c-myc gene. Nucleic Acids Res. 2002, 30, 2701–2709. [Google Scholar] [PubMed]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Watanabe, T.; Kimura, M.T.; Koshikawa, N.; Ikeda, M.; Uekusa, S.; Kawashima, H.; Wang, X.; Igarashi, J.; Choudhury, D.; et al. Identification of a novel E-box binding pyrrole-imidazole polyamide inhibiting MYC-driven cell proliferation. Cancer Sci. 2015, 106, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin. Cancer Res. 2016, 22, 5539–5552. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of STAT3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. The JAK2 inhibitor AZD1480 potently blocks STAT3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar]
- Fizazi, K.; De Bono, J.S.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.B.; Qi, M.; Bandekar, R.; Vermeulen, J.T.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, B.; Eisenberger, M.A.; Rettig, M.B.; Chu, F.; Pili, R.; Stephenson, J.J.; Vogelzang, N.J.; Koletsky, A.J.; Nordquist, L.T.; Edenfield, W.J.; et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Centenera, M.M.; Gillis, J.L.; Hanson, A.R.; Jindal, S.; Taylor, R.A.; Risbridger, G.P.; Sutherland, P.D.; Scher, H.I.; Raj, G.V.; Knudsen, K.E.; et al. Evidence for Efficacy of New Hsp90 Inhibitors Revealed by Ex Vivo Culture of Human Prostate Tumors. Clin. Cancer Res. 2012, 18, 3562–3570. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Taylor, R.A.; Toivanen, R.; Pedersen, J.; Norden, S.; Pook, D.W.; Frydenberg, M.; Australian Prostate Cancer, B.; Papargiris, M.M.; Niranjan, B.; et al. A preclinical xenograft model of prostate cancer using human tumors. Nat. Protoc. 2013, 8, 836–848. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Factor | Functions for AR | Efficacy for Cancer Progression | FOXA1 Interaction | Related Drugs | Reference |
|---|---|---|---|---|---|
| FOXA1 | Pioneer factor | Controversial | GSK126 | [141] | |
| GATA2 | Pioneer factor/Activator | Promote | + | K-7174 | [143,145,146,147] |
| OCT1 | Activator | Promote | + | PI polyamide | [72] |
| ETS1 | Activator | Promote | − | ODNs | [149,150] |
| ERG | Repressor | Promote | − | PI polyamide/YK-4-279/DB1255/WP1130 | [151,153,154,155] |
| NKX3-1 | Activator | Controversial | + | RNAa | [157] |
| C/EBPs | Repressor | Unknown | – | RNAa | [159] |
| NFI | Diverse effects on gene regulation | Unknown | + | - | |
| RUNX1 | Activator | Inhibit | − | - | |
| FOXP1 | Repressor | Inhibit | + | - | |
| E2F | Activator (CRPC) | Promote | − | Thymoquinone/Peptide | [160,161,162] |
| MYC | Controversial (CRPC) | Promote | − | CX5461/CX6258 | [167] |
| STAT3 | Activator (CRPC) | Controversial | − | ODNs/AZD1480/Siltuximab | [168,169,170] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers 2017, 9, 22. https://doi.org/10.3390/cancers9030022
Obinata D, Takayama K, Takahashi S, Inoue S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers. 2017; 9(3):22. https://doi.org/10.3390/cancers9030022
Chicago/Turabian StyleObinata, Daisuke, Kenichi Takayama, Satoru Takahashi, and Satoshi Inoue. 2017. "Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer" Cancers 9, no. 3: 22. https://doi.org/10.3390/cancers9030022
APA StyleObinata, D., Takayama, K., Takahashi, S., & Inoue, S. (2017). Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers, 9(3), 22. https://doi.org/10.3390/cancers9030022