
MTH1 as a Chemotherapeutic Target: The Elephant in the Room


Abstract
:1. Introduction
2. A Functional Role for MTH1 in Tumor Biology
3. Outcomes of MTH1 Inhibitors in Different Cancer Models
4. Molecular and Cellular Contexts Underlying the Outcomes of MTH1 Inhibition
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Mo, J.Y.; Maki, H. Molecular mechanisms for controlling spontaneous and induced mutagenesis. Nucleic Acids Symp. Ser. 1992, 27, 101–102. [Google Scholar]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001, 477, 59–70. [Google Scholar] [CrossRef]
- Sakai, Y.; Furuichi, M.; Takahashi, M.; Mishima, M.; Iwai, S.; Shirakawa, M.; Nakabeppu, Y. A molecular basis for the selective recognition of 2-hydroxy-dATP and 8-oxo-dGTP by human MTH1. J. Biol. Chem. 2002, 277, 8579–8587. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Tsuzuki, T. Oxidative nucleotide damage: Consequences and prevention. Oncogene 2002, 21, 8895–8904. [Google Scholar] [CrossRef] [PubMed]
- Kamath-Loeb, A.S.; Hizi, A.; Kasai, H.; Loeb, L.A. Incorporation of the guanosine triphosphate analogs 8-oxo-dGTP and 8-NH2-dGTP by reverse transcriptases and mammalian DNA polymerases. J. Biol. Chem. 1997, 272, 5892–5898. [Google Scholar] [CrossRef] [PubMed]
- Katafuchi, A.; Nohmi, T. DNA polymerases involved in the incorporation of oxidized nucleotides into DNA: Their efficiency and template base preference. Mutat. Res. 2010, 703, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Einolf, H.J.; Guengerich, F.P. Fidelity of nucleotide insertion at 8-oxo-7,8-dihydroguanine by mammalian DNA polymerase delta. Steady-state and pre-steady-state kinetic analysis. J. Biol. Chem. 2001, 276, 3764–3771. [Google Scholar] [CrossRef] [PubMed]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef] [PubMed]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chaudhry, M.A.; Wallace, S.S. Base excision repair by hNTH1 and hOGG1: A two edged sword in the processing of DNA damage in gamma-irradiated human cells. DNA Repair 2006, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, H.; Hofer, A.; Thelander, L.; Kitajima, S.; Cai, Y.; Oshiro, S.; Yakushiji, H.; Nakabeppu, Y.; Kuwano, M.; Sekiguchi, M. Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 1999, 38, 3610–3614. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D. 8-Oxo-deoxyguanosine: Reduce, reuse, recycle? Proc. Natl. Acad. Sci. USA 2007, 104, 13535–13536. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Kamiya, H.; Yakushiji, H.; Fujii, Y.; Nakabeppu, Y.; Kasai, H. The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J. Biol. Chem. 1999, 274, 18201–18205. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, D.; Sakumi, K.; Ohno, M.; Sakai, Y.; Furuichi, M.; Iwai, S.; Nakabeppu, Y. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. Biol. Chem. 2003, 278, 37965–37973. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Coskun, E.; Jaruga, P.; Jemth, A.S.; Loseva, O.; Scanlan, L.D.; Tona, A.; Lowenthal, M.S.; Helleday, T.; Dizdaroglu, M. Addiction to MTH1 protein results in intense expression in human breast cancer tissue as measured by liquid chromatography-isotope-dilution tandem mass spectrometry. DNA Repair 2015, 33, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.H.; Pass, H.I.; Mitchell, J.B. Expression of human MutT homologue (hMTH1) protein in primary non-small-cell lung carcinomas and histologically normal surrounding tissue. Free Radic. Biol. Med. 2003, 34, 1447–1457. [Google Scholar] [CrossRef]
- Rai, P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: Defective bricks build a defective house. Mutat. Res. 2010, 703, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wikman, H.; Risch, A.; Klimek, F.; Schmezer, P.; Spiegelhalder, B.; Dienemann, H.; Kayser, K.; Schulz, V.; Drings, P.; Bartsch, H. hOGG1 polymorphism and loss of heterozygosity (LOH): Significance for lung cancer susceptibility in a caucasian population. Int. J. Cancer 2000, 88, 932–937. [Google Scholar] [CrossRef]
- Mambo, E.; Chatterjee, A.; de Souza-Pinto, N.C.; Mayard, S.; Hogue, B.A.; Hoque, M.O.; Dizdaroglu, M.; Bohr, V.A.; Sidransky, D. Oxidized guanine lesions and hOgg1 activity in lung cancer. Oncogene 2005, 24, 4496–4508. [Google Scholar] [CrossRef] [PubMed]
- Speina, E.; Arczewska, K.D.; Gackowski, D.; Zielinska, M.; Siomek, A.; Kowalewski, J.; Oliński, R.; Tudek, B.; Kuśmierek, J.T. Contribution of hMTH1 to the Maintenance of 8-Oxoguanine Levels in Lung DNA of Non-Small-Cell Lung Cancer Patients. J. Natl. Cancer Inst. 2005, 97, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Rai, P. MTH1 expression is required for effective transformation by oncogenic HRAS. Oncotarget 2015, 6, 11519–11529. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Burton, D.G.; Halvorsen, K.; Balkan, W.; Reiner, T.; Perez-Stable, C.; Cohen, A.; Munoz, A.; Giribaldi, M.G.; Singh, S. MutT Homolog 1 (MTH1) maintains multiple KRAS-driven pro-malignant pathways. Oncogene 2015, 34, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Egashira, A.; Yamauchi, K.; Yoshiyama, K.; Kawate, H.; Katsuki, M.; Sekiguchi, M.; Sugimachi, K.; Maki, H.; Tsuzuki, T. Mutational specificity of mice defective in the MTH1 and/or the MSH2 genes. DNA Repair 2002, 1, 881–893. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Ichinoe, A.; Ohno, M.; Ide, Y.; Hirano, S.; Yoshimura, D.; Tominaga, Y.; Furuichi, M.; Sakumi, K. Biological significance of the defense mechanisms against oxidative damage in nucleic acids caused by reactive oxygen species: From mitochondria to nuclei. Ann. N. Y. Acad. Sci. 2004, 1011, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Onder, T.T.; Young, J.J.; McFaline, J.L.; Pang, B.; Dedon, P.C.; Weinberg, R.A. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. Natl. Acad. Sci. USA 2009, 106, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Young, J.J.; Burton, D.G.; Giribaldi, M.G.; Onder, T.T.; Weinberg, R.A. Enhanced elimination of oxidized guanine nucleotides inhibits oncogenic RAS-induced DNA damage and premature senescence. Oncogene 2011, 30, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Rai, P. Human Mut T Homolog 1 (MTH1): A roadblock for the tumor-suppressive effects of oncogenic RAS-induced ROS. Small GTPases 2012, 3, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Rai, P. MTH1 counteracts oncogenic oxidative stress. Oncoscience 2015, 2, 785–786. [Google Scholar] [PubMed]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H.; et al. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521. [Google Scholar] [CrossRef] [PubMed]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and Selective Inhibitors of MTH1 Probe Its Role in Cancer Cell Survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef] [PubMed]
- Petrocchi, A.; Leo, E.; Reyna, N.J.; Hamilton, M.M.; Shi, X.; Parker, C.A.; Mseeh, F.; Bardenhagen, J.P.; Leonard, P.; Cross, J.B.; et al. Identification of potent and selective MTH1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Chow, A.S.; Au, J.S. MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol. 2011, 8, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Warpman Berglund, U.; Sanjiv, K.; Gad, H.; Kalderen, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Clarkin, K.C.; Wahl, G.M. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc. Natl. Acad. Sci. USA 1996, 93, 4827–4832. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.; Taxman, D.J. Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003, 5, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species- and cell type-specific requirements for cellular transformation. Cancer Cell 2004, 6, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, M.; Horton, J.K.; Dai, D.P.; Stefanick, D.F.; Wilson, S.H. Oxidized nucleotide insertion by pol beta confounds ligation during base excision repair. Nat. Commun. 2017, 8, 14045. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, L.; Pudelko, L.; Jemth, A.S.; Gad, H.; Narwal, M.; Gustafsson, R.; Karsten, S.; Puigvert, J.C.; Homan, E.; Berndt, C.; et al. Hypoxic Signaling and the Cellular Redox Tumor Environment Determine Sensitivity to MTH1 Inhibition. Cancer Res. 2016, 76, 2366–2375. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Mistry, V.; Singh, R.; Gackowski, D.; Rozalski, R.; Siomek-Gorecka, A.; Phillips, D.H.; Zuo, J.; Mullenders, L.; Pines, A.; et al. Nucleotide excision repair of oxidised genomic DNA is not a source of urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine. Free Radic. Biol. Med. 2016, 99, 385–391. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Kenneth, H.Y.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]



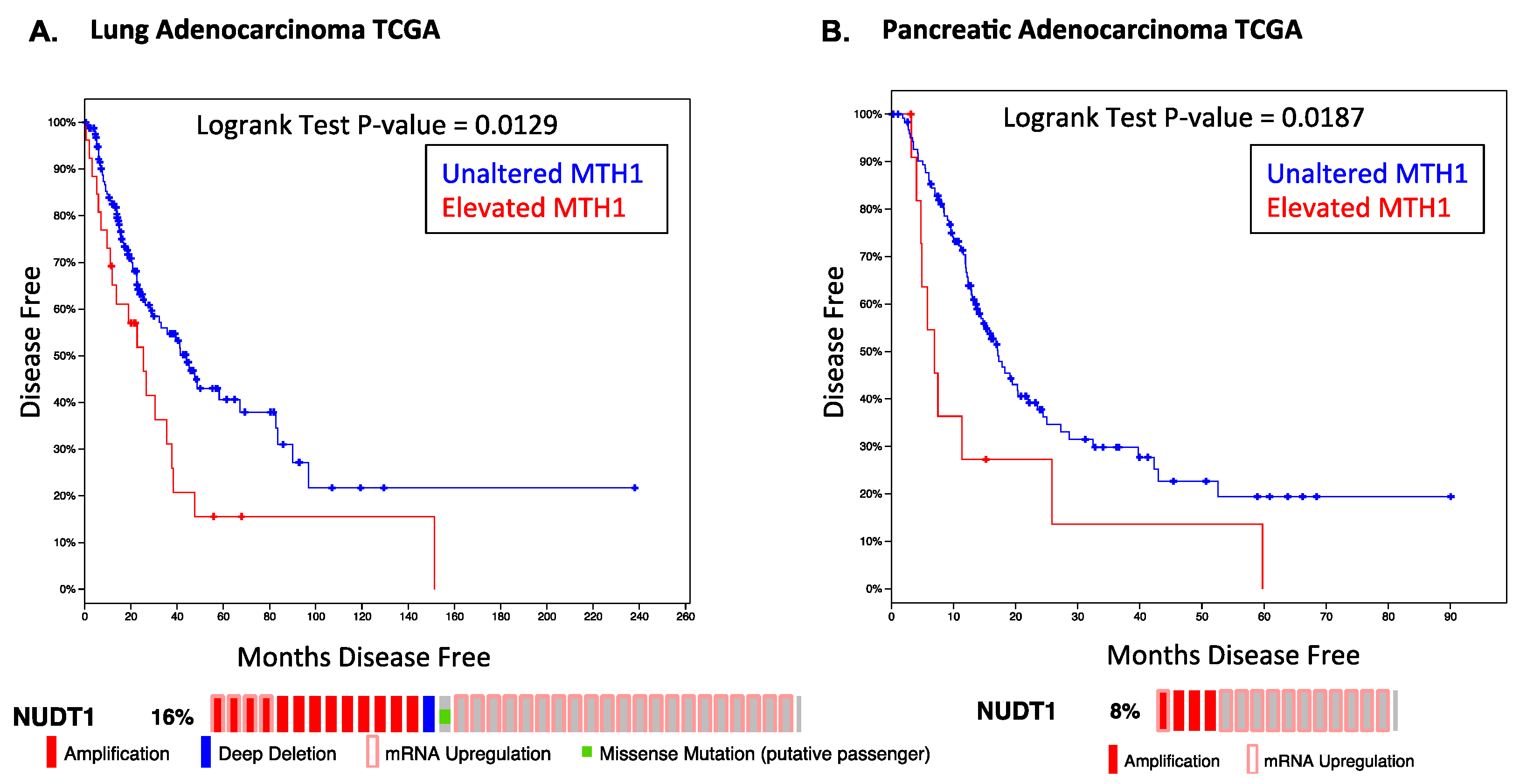{kind=link}
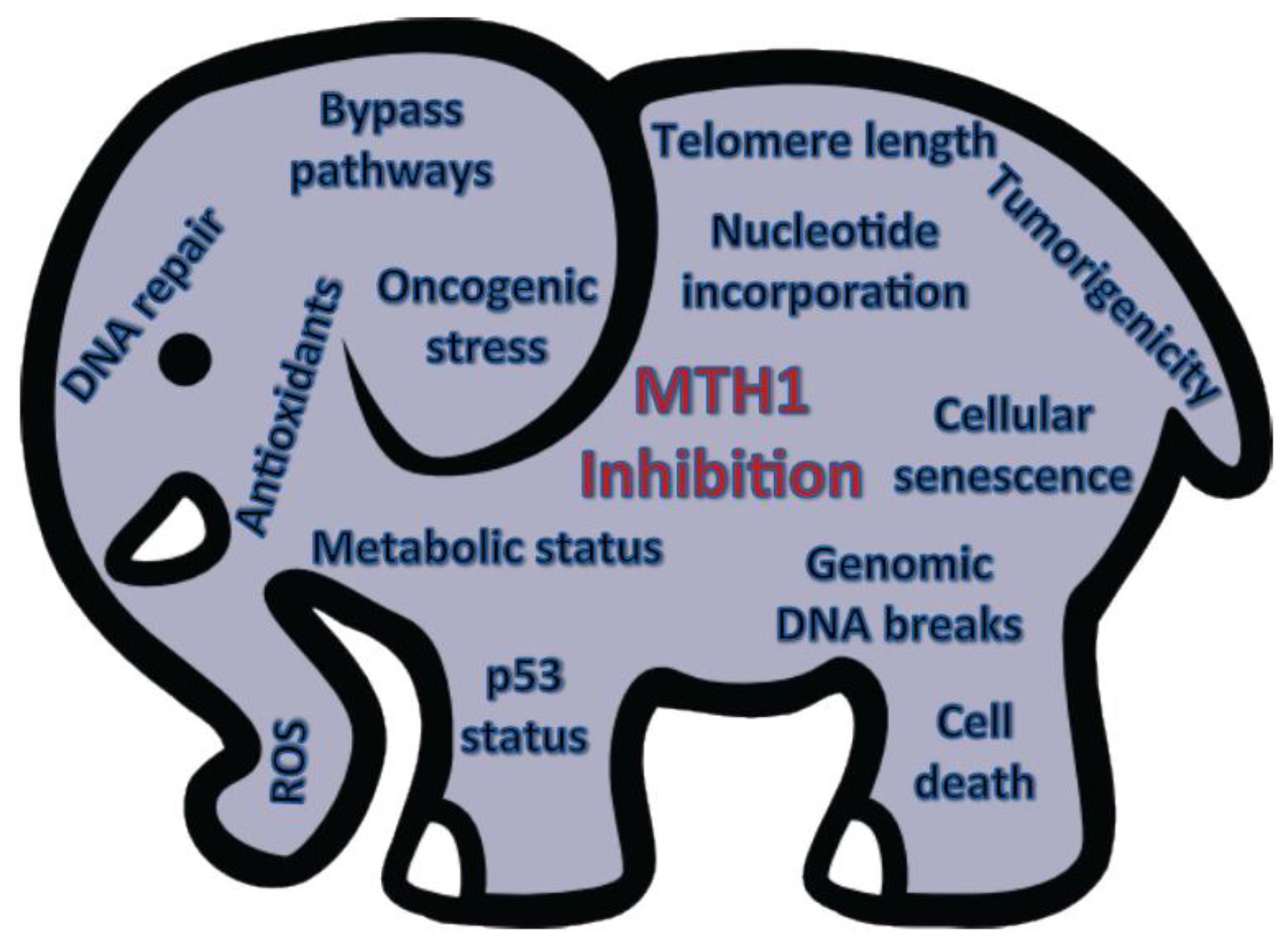{kind=link}
| Publication | Cell Line | Cell Type | Mode of MTH1 Inhibition | Mode of Measurement | Outcome |
|---|---|---|---|---|---|
| Gad et al., Nature, 2014 [35] | U2OS | Human osteosarcoma | siRNA, TH287, TH588 | -Cell viability, clonogenic survival -Immunofluorescent staining (8-oxoGua via avidin-AlexaFluor488 staining , 53BP1, RPA, phospho-S1981 ATM, RAD51, phospho-S2056 DNA-PKcs, cleaved caspase 3, H3K9me3) -OGG1/MUTYH alkaline comet assay -Immunoblot p53 (S15), p21cip1, phospho-S1981 ATM | -siRNA against MTH1: decreased cell viability and clonogenic survival, increased total cellular 8-oxoGua, RPA, 53BP1 foci, H3K9me3, and cleaved caspase 3. DNA damage was induced as measured by RAD51, DNA-PKcs mediated DSB repair, increased ATM-dependent (S15) phosphorylation, p21cip1. Increased OGG1/MUTYH comet tail moment showing increased 8-oxo-dG incorporation into DNA -TH588 or TH287: increased total cellular 8-oxoGua, elevated cytotoxicity, DNA damage (induced 53BP1, DNA-PKcs foci, increased OGG1/MUTYH comet tail moment showing increased 8-oxo-dG incorporation into DNA) |
| VH10 | Primary cells, human foreskin fibroblasts | siRNA, TH287, TH588 | -Cell viability, clonogenic survival -Immunofluorescent staining (8-oxoGua via avidin-AlexaFluor488 staining , 53BP1, RPA, phospho-DNA-PKcs) -Immunoblot (p53 (S15), p21cip1, phospho-S1981 ATM | -No decrease in clonogenic survival nor increase in total cellular 8-oxoGua with MTH1 siRNA -No 53BP1, RPA, DNA-PKcs foci with TH287, TH588 -No change in ATMpS1981, p53 (S15), or p21cip1 protein levels with TH287 | |
| BJ cells with hTERT, hTERT/SV40LgT or hTERT/SV40LgT/RasV12 | Human foreskin fibroblasts | TH287, TH588 | -Cell viability and clonogenic survival | -Reduced cell viability and clonogenic survival in hTERT/SV40T and hTERT/SV40T/RasV12 cells, compared to hTERT cells | |
| HCT116 WT or p53 −/− | Human colorectal cancer | TH287, TH588 | -Cell viability | -No significant difference in viability between isogenic p53 WT and p53 −/− cells | |
| Patient Derived Xenograft | Metastases from patient with BRAFV600E-mutated melanoma, whose tumor was resistant to carboplatin/dacarbazine/vemurafenib | TH588, once daily, 30 mg/kg, for 20 days | -In vivo: TH588 subcutaneously injected in female NOD-SCID IL2Rγnull (NOG) mice, tumor growth monitored | -Significant reduction in tumor growth | |
| MCF7 | Human breast adenocarcinoma | TH588, once daily, 30 mg/kg, for 18 days | -In vivo mouse xenograft: tumor volume measured | -Significant reduction in tumor volume | |
| SW480 | Human colorectal cancer cell line (established from primary tumor) | -In vitro: siRNA #3, dox-inducible shMTH1, TH588 -In vivo: Dox inducible shMTH1 cells, TH588, once daily, 30 mg/kg, for 35 days | -Cell survival -Subcutaneous xenograft tumors in female SCID mice | -Reduced cell survival -Reduced mouse xenograft tumors, improved survival | |
| SW620 | Human colorectal cancer cell line (from metastatic SW480 tumor) | siRNA # 3 | -Cell survival | -Better survival under MTH1 siRNA vs. SW480 cells | |
| HeLa | Human cervical cancer | siRNA # 3 | -Cell survival | -Low surviving fraction in siMTH1 cells | |
| HEK293T | Human embryonic kidney, SV40 T-antigen | siRNA # 3 | -Cell survival | -Low surviving fraction in siMTH1 cells | |
| MB231 | Human breast adenocarcinoma | siRNA # 3 | -Cell survival | -Intermediate surviving fraction in siMTH1 cells | |
| LNCaP, DU145, PC-3 | Human prostate cancer | siRNA # 3 | -Cell survival | -Better survival in siMTH1 LNCaP and PC-3 cells vs. DU145 | |
| Huber et al., Nature, 2014 [36] | SW480 | Human colorectal cancer | SCH51344, (S)-Crizotinib, shRNA, siRNA | -Viability, colony formation -Immunofluorescent staining for DNA damage markers: 53BP1 and phospho-S1981 ATM -MUTYH/OGG1 alkaline comet assay -SCID xenograft tumor model | -SCH51344 and (S)-Crizotinib decreased cell viability, increased 53BP1 and ATM autophosphorylation, increased comet tail moments. MTH1 overexpression reduced DSBs -MTH1 siRNA and shRNA impaired colony formation -No difference in viability between ATM or ATR inhibitor-treated shMTH1 and shGFP -(S)-Crizotinib significantly reduced xenograft tumors |
| SW480 shp53 Tet-on system | Human colorectal cancer | SCH51344, (S)-Crizotinib | -Viability | -No significant difference in viability under p53 knockdown | |
| HCT116 WT, p53 −/−, p21 −/− | Human colorectal cancer | SCH51344, (S)-Crizotinib | -Viability | -SCH51344: no difference in viability between WT, p53 −/−, and p21 −/− cells -(S)-Crizotinib: p21 −/− cells were strongly sensitized compared to WT and p53 −/− cells | |
| BJ cells: WT, hTERT, hTERT/SV40Lg T, or hTERT/SV40LgT/RasV12 | Human foreskin fibroblasts | SCH51344, (S)-Crizotinib | -Viability | -Reduced cell viability in hTERT/SV40T and hTERT/SV40T/RasV12 cells, compared to hTERT or WT cells -Reduced viability with (S)-Crizotinib vs. SCH51344 | |
| DLD1 | Human colorectal cancer | siRNA | -Colony formation | -siMTH1 impaired colony formation | |
| ATM proficient vs. ATM −/− MEFs | Mouse embryonic Fibroblasts (MEFs) | SCH51344, (S)-Crizotinib | -Viability | -No difference in viability between ATM −/− and ATM-proficient MEFs | |
| PANC-1 | Human pancreatic cancer | (S)-Crizotinib | -Colony formation | -Inhibition of colony formation | |
| Warpman et al., Annals of Oncology 2016 [42] | U2OS | Human osteosarcoma | siRNA, TH1579 | -Cell viability -Modified comet assay (with MutT, or OGG1, or N-acetyl cysteine) -Immunofluorescence for DNA damage foci: DNA-PK, XRCC1, γH2AX, 53BP1 | -TH1579 effectively introduced 8-oxoGua into cells, and this effect was reversed with N-acetyl cysteine, overexpression of human MTH1 or bacterial MutT. Effect can be enhanced by treatment with OGG1. Increased DNA damage foci i.e., DNA-PK, XRCC1, γH2AX, 53BP1 was observed. |
| SW480 | Human colorectal cancer | TH1579, in vitro and in vivo. For in vivo model: once daily at 30 mg/kg, 60 mg/kg, or 90 mg/kg | -In vitro: Modified comet assay -In vivo: Xenograft mouse model, tumor volume and animal weight measured | -MTH1 overexpression mitigated comet taillength as measured by a modified comet assay, following TH1579 treatment. Comet tail moment increased with OGG1 treatment -Effective reduction in tumor growth | |
| SW480.SN3: derivative of SW480 | Human colorectal cancer | shRNA | -In vitro: Cell viability -In vivo: injected doxycycline-inducible shMTH1 in female SCID mice, tumor volumes were measured | -Tumor formation in mice reflected/tracked with level of MTH1 knockdown in vitro | |
| BJ hTERT/SV40T/RasV12 | Transformed human foreskin fibroblasts | TH1579 | -Cell viability | -More cytotoxicity in BJ-hTERT-Ras-SV40T cells vs. non-transformed counterpart (BJ-hTERT) | |
| NTUB1/P | Human drug resistant bladder cancer | -TH588, TH816, IACS-4759, TH1579 -AstraZeneca (AZ) compounds 15, 19, 24 [38] -AZ siRNA [38] | -Chemical inhibitors of MTH1: 8-oxo-dG incorporation via immunofluorescence -AZ siRNA: Cell viability | -8-oxo-dG incorporation into the DNA correlated with MTH1-inhibition associated cytotoxicity -AZ siRNA: Resulted in loss of viability in NTUBP1/P cancer cells | |
| Patient Derived Xenograft | Metastatic carboplatin/dacarbazine/vemurafenib –resistant BRAFV600E-mutated melanoma, | TH1579, in vivo: 45 mg/kg daily, via oral gavage for approximately 40 days | -Tumor growth measured in mice | -Significant reduction in tumor growth | |
| HCT116 | Human colorectal cancer | TH588, TH1579 | -TH588 and anti-tubulin drugs: Proteomics profile -TH1579 in vitro: Immunoblot to assess phospho-p53 (S15), total p53, p21, cleaved PARP, cleaved caspase 3, γH2AX -TH1579 in vivo: Mouse xenograft experiment, 90 mg/kg. Measured 8-oxo-dG, number of 53BP1, γH2AX, caspase 3, Ki67 foci. Immunoblotting of mouse tumors for phospho-p53 (S15), total p53, p21, cleaved PARP, γH2AX levels | -Proteomics profile: TH588 clustered with nutrient starvation—Immunoblot in vitro TH1579 treatment: increased phospho-p53, total p53, p21, cleaved PARP, cleaved caspase 3, and γH2AX -In vivo TH1579 treatment: increased 8-oxo-dG, 53BP1 and caspase 3 foci. A decrease in Ki67 foci was observed. Immunoblot: elevated p53, small increase in phospho-p53, increase in p21, minor elevation in cleaved PARP levels, no noticeable change in γH2AX levels | |
| Kawamura et al., Scientific Reports 2016 [37] | HeLa | Human cervical cancer | NPD7155, NPD9948, TH287, (S)-Crizotinib, SCH51344, MTH1 siRNA | -Cell viability -8-oxo-dG and 53BP1 foci measured via immunofluorescence -Proteomics profiling and tubulin polymerization -Cell cycle analysis -Immunoblot for Bcl-2 | -NPD7155 and NPD9948 exhibited weak cytotoxicity, compared to TH287 and (S)-Crizotinib. NPD7155 and NPD9948 did not introduce 8-oxo-dG into DNA to the same extent as TH287. NPD7155 and NPD9948 induced 53BP1 foci at 100 µM -TH287 and TH588 inhibited tubulin polymerization, at 30 µM or higher -TH287 and TH588, but not NPD7155 and NPD9948, increased cells in G2/M phase, -TH287 and TH588 induced mild Bcl-2 phosphorylation, similar to tubulin-targeting agent Vinblastine (immunoblot for supershift in Bcl2 signal) -MTH1 siRNA did not affect cell survival or cell cycle progression |
| PANC-1 | Human pancreatic cancer | NPD7155, NPD9948, TH287, (S)-Crizotinib, SCH51344 | -Cell viability | -100 µM, NPD7155: ~ 50% loss of viability -100 µM NPD9948: ~ 30% loss of viability -TH287: 10 µM, 70% reduction in viability -(S)-Crizotinib: 10 µM, ~20% reduction in viability -SCH51344: 10 µM, ~15% reduction in viability | |
| MIA PaCa-2 | Human pancreatic cancer | NPD7155, NPD9948, TH287, (S)- Crizotinib, SCH51344 | -Cell viability | -30 µM, NPD7155: ~25% loss of viability -30 µM, NPD9948: no loss of viability -TH287: 3 µM, 90% reduction in viability -(S)-Crizotinib: 10 µM, 70% reduction in viability -SCH51344: 10 µM, 30% reduction in viability | |
| NIH3T3 or NIH3T3/KRAS | Non-transformed MEFs, vs. KRAS transformed MEFs | NPD7155, NPD9948, TH287, (S)-Crizotinib, SCH51344 | -Cell viability | -NPD7155: Small decrease in viability in NIH3T3/KRAS compared to NIH3T3 cells -Remaining MTH1 inhibitors: no significant difference in viability between NIH3T3/KRAS and NIH3T3 cells | |
| MG-63 | Human osteosarcoma | NPD7155, NPD9948, TH287, (S)-Crizotinib, SCH51344 | -Cell viability | -30 µM, NPD7155: ~20% loss of viability -30 µM NPD9948: 10% loss of viability -TH287: 3 µM, 90% reduction in viability -(S)-Crizotinib: 10 µM, 60% reduction in viability -SCH51344: 10 µM, ~25% reduction in viability | |
| Kettle et al., Journal of Medicinal Chemistry, 2016 [38] | U2OS, A549, H358, MCF7 | Human osteosarcoma, lung adenocarcinomas, breast adenocarcinoma | TH588, S-crizotinib, AZ compounds 15, 19 | -Growth inhibition (GI50) | -TH588 and (S)-Crizotinib had low GI50s, in the range of 2 to 5 µM. Compounds 15 and 19 had high GI50s: >30 µM for compound 15, and between 6 and 14 µM for compound 19 |
| U2OS | Human osteosarcoma | TH588, compound 19, commercially available siRNA, siRNA # 3 from Gad et al., 2014 [35] | -TH588, compound 19: DNA damage response (DDR) signaling markers via immunoblot: p-Ser1981 ATM, p-Ser15 p53, γH2AX, RPA, cleaved PARP1 -Cell viability | -Elevated p-Ser15p53, total p53, cleaved-PARP1 with TH588. No effect on DDR signaling activation or apoptosis with compound 19. Neither compound changed γH2AX or RPA levels. -Commercially available siRNA did not affect cell viability, siRNA # 3 strongly impaired cell viability -TH588, (S)-Crizotinib, and compound 19 killed MTH1 siRNA transfected cells | |
| SW480 | Human colorectal cancer | CRISPR-mediated knockout | -Cell viability | -Clones with complete MTH1 knockout did not show impaired growth relative to parental MTH1 WT cells -(S)-Crizotinib and compound 19 killed MTH1 knockout and parental cells equivalently | |
| Petrocchi et al., Bioorg. Med. Chem. Lett., 2016 [39] | U2OS, SaOS2, SW480, MDA-MB-231, HeLa, UOK262, 293T, A549, H460, H358, WI38, BJ, hMEC | Human cell lines (osteosarcoma, colorectal/breast/cervical cancer, embryonic kidney, lung cancer, fetal lung and foreskin fibroblasts, mammary epithelial | IACS-4759, IACS-4619 | -Cell viability | -No cytotoxicity at concentrations up to 50 µM. Data not shown |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samaranayake, G.J.; Huynh, M.; Rai, P. MTH1 as a Chemotherapeutic Target: The Elephant in the Room. Cancers 2017, 9, 47. https://doi.org/10.3390/cancers9050047
Samaranayake GJ, Huynh M, Rai P. MTH1 as a Chemotherapeutic Target: The Elephant in the Room. Cancers. 2017; 9(5):47. https://doi.org/10.3390/cancers9050047
Chicago/Turabian StyleSamaranayake, Govindi J., Mai Huynh, and Priyamvada Rai. 2017. "MTH1 as a Chemotherapeutic Target: The Elephant in the Room" Cancers 9, no. 5: 47. https://doi.org/10.3390/cancers9050047
APA StyleSamaranayake, G. J., Huynh, M., & Rai, P. (2017). MTH1 as a Chemotherapeutic Target: The Elephant in the Room. Cancers, 9(5), 47. https://doi.org/10.3390/cancers9050047