
Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma

Abstract
:1. Glioblastoma
1.1. HGF and MET
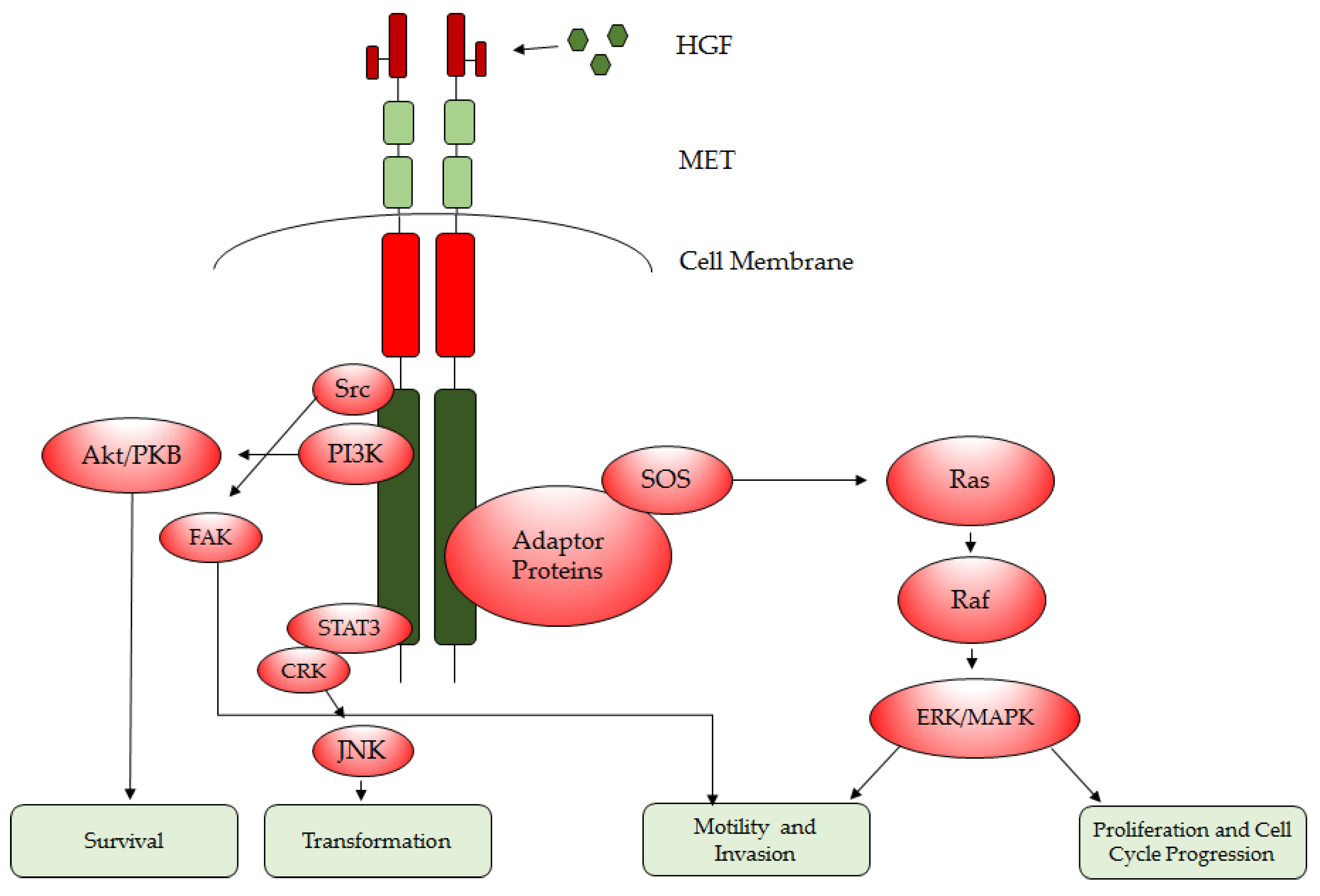
1.2. HGF/MET Downstream Signaling
1.3. HGF/MET Regulation by microRNAs
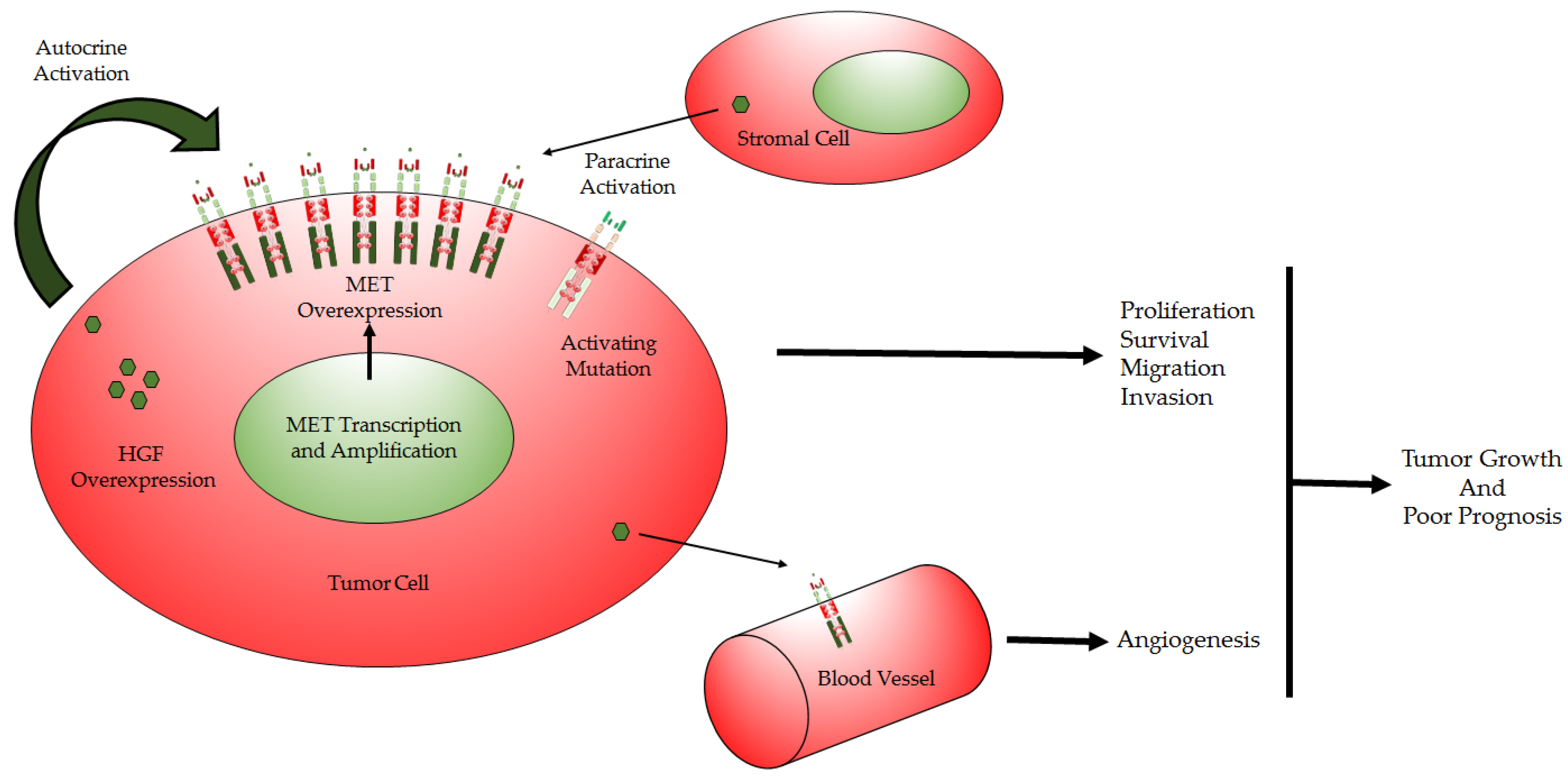
1.4. HGF/MET Deregulation in GBM
1.5. HGF/MET Functions and Modes of Action in GBM
1.5.1. Cell Cycle Regulation
1.5.2. Cell Proliferation and Evasion of Apoptosis
1.5.3. Cell Migration and Invasion
1.5.4. Angiogenesis
1.5.5. Glioblastoma Stem Cells and Therapy Resistance
1.6. HGF/MET as GBM Therapeutic Targets
1.6.1. Competitors of MET (Preclinical)
1.6.2. Small-Molecule Inhibitors (Preclinical)
1.6.3. Small-Molecule Inhibitors (Clinical)
1.6.4. Monoclonal Antibodies against MET and HGF
2. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. Onco. Targets Ther. 2017, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, AND NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; DiPierro, C.; Zhang, Y.; Mudrick, T.; Fuller, L.; Johnson, E.; Marcinkiewicz, L.; Engelhardt, M.; Kefas, B.; et al. Interactions between PTEN and the c-MET pathway in glioblastoma and implications for therapy. Mol. Cancer Ther. 2009, 8, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Awad, A.J.; Burns, T.C.; Zhang, Y.; Abounader, R. Targeting MET for glioma therapy. Neurosurg. Focus 2014, 37, E10. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Garajova, I.; Giovannetti, E.; Biasco, G.; Peters, G.J. c-MET as a target for personalized therapy. Transl. Oncogenomics 2015, 7, 13–31. [Google Scholar] [PubMed]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro. Oncol. 2005, 7, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed]
- Navis, A.C.; van Lith, S.A.; van Duijnhoven, S.M.; de Pooter, M.; Yetkin-Arik, B.; Wesseling, P.; Hendriks, W.J.; Venselaar, H.; Timmer, M.; van Cleef, P.; et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. 2015, 130, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Farenholtz, K.E.; Yang, Y.; Guessous, F.; Dipierro, C.G.; Calvert, V.S.; Deng, J.; Schiff, D.; Xin, W.; Lee, J.K.; et al. Hepatocyte growth factor sensitizes brain stumors to c-MET kinase inhibition. Clin. Cancer Res. 2013, 19, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jain, R.K.; Zhu, M. Recent progress and advances in HGF/MET-targeted therapeutic agents for cancer treatment. Biomedicines 2015, 3, 149–181. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Graziani, A. Physiological signaling and structure of the HGF receptor MET. Biomedicines 2015, 3, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/MET signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Gozdzik-Spychalska, J.; Szyszka-Barth, K.; Spychalski, L.; Ramlau, K.; Wojtowicz, J.; Batura-Gabryel, H.; Ramlau, R. c-MET inhibitors in the treatment of lung cancer. Curr. Treat. Options Oncol. 2014, 15, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Petrini, I. Biology of MET: A double life between normal tissue repair and tumor progression. Ann. Transl. Med. 2015, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.G.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc. Natl. Acad. Sci. USA 2006, 103, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.; Feng, Y.; Li, R.; Sun, X.; Du, W.; Piao, X.; Wang, H.; Yang, D.; Sun, Y.; et al. miR-410 regulates MET to influence the proliferation and invasion of glioma. Int. J. Biochem. Cell Biol. 2012, 44, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Yu, H.; Hu, M.; Xia, T.; Yue, X. miR-144-3p exerts anti-tumor effects in glioblastoma by targeting c-MET. J. Neurochem. 2015, 135, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Zhen, L.; Yun-Hui, L.; Hong-Yu, D.; Jun, M.; Yi-Long, Y. Long noncoding RNA NEAT1 promotes glioma pathogenesis by regulating miR-449b-5p/c-MET axis. Oncodev. Biol. Med. 2016, 37, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Nguyen, D.H.; Dong, Q.; Shitaku, P.; Chung, K.; Liu, O.Y.; Tso, J.L.; Liu, J.Y.; Konkankit, V.; Cloughesy, T.F.; et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J. Neurooncol. 2009, 94, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kouri, F.M.; Hurley, L.A.; Daniel, W.L.; Day, E.S.; Hua, Y.; Hao, L.; Peng, C.Y.; Merkel, T.J.; Queisser, M.A.; Ritner, C.; et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015, 29, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, J.; Mueller, A.C.; Dey, B.; Yang, Y.; Lee, D.H.; Hachmann, J.; Finderle, S.; Park, D.M.; Christensen, J.; et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014, 21, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. The hepatocyte growth factor receptor: Structure, function and pharmacological targeting in cancer. Curr. Signal Transduct. Ther. 2011, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Kataoka, H. Mechanisms of hepatocyte growth factor activation in cancer tissues. Cancers 2014, 6, 1890–1904. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Kataoka, H.; Koono, M.; Wakisaka, S. Expression of hepatocyte growth factor/scatter factor and its receptor c-MET in brain tumors: Evidence for a role in progression of astrocytic tumors (review). Int. J. Mol. Med. 1999, 3, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Invest. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.A.; Hossain, M.A.; Luddy, C.; Goel, N.; Reznik, T.E.; Laterra, J. Scatter factor/hepatocyte growth factor stimulation of glioblastoma cell cycle progression through G(1) is c-MYC dependent and independent of p27 suppression, CDK2 activation, or E2f1-dependent transcription. Mol. Cell Biol. 2002, 22, 2703–2715. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-MET as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, H.C. Direct interaction of focal adhesion kinase (FAK) with met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; McFarland, B.C.; Nozell, S.E.; Benveniste, E.N. NF-kappaB and STAT3 in glioblastoma: Therapeutic targets coming of age. Expert Rev. Neurother. 2014, 14, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, T.; Ma, P.; Mitteer, R.A., Jr.; Zhang, Z.; Kim, H.J.; Yeo, E.; Zhang, D.; Cai, P.; Li, C.; et al. C-MET-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Invest. 2016, 126, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- McCarty, J.H. Glioblastoma resistance to anti-VEGF therapy: Has the challenge been MET? Clin. Cancer Res. 2013, 19, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Joo, K.M.; Jin, J.; Nam, D.H. Cancer stem cells and their mechanism of chemo-radiation resistance. Int. J. Stem Cells 2009, 2, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.M.; Jin, J.; Kim, E.; Ho Kim, K.; Kim, Y.; Gu Kang, B.; Kang, Y.J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cazares, H.; Eberhart, C.G.; et al. c-MET signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med. 2016, 8, 550–568. [Google Scholar] [CrossRef] [PubMed]
- Koso, H.; Takeda, H.; Yew, C.C.; Ward, J.M.; Nariai, N.; Ueno, K.; Nagasaki, M.; Watanabe, S.; Rust, A.G.; Adams, D.J.; et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl. Acad Sci. USA 2012, 109, E2998–E3007. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Fujita, M.; Okuda, T.; Yoneshige, A.; Nakata, S.; Yamashita, K.; Yoshioka, H.; Izumoto, S.; Kato, A. MET expressed in glioma stem cells is a potent therapeutic target for glioblastoma multiforme. Anticancer Res. 2016, 36, 3571–3577. [Google Scholar] [PubMed]
- Rath, P.; Lal, B.; Ajala, O.; Li, Y.; Xia, S.; Kim, J.; Laterra, J. In vivo c-MET pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Transl. Oncol. 2013, 6, 104–111. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D’Ambrosio, A.; Porrati, P.; et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Seol, H.J.; Kim, E.H.; Rheey, J.; Jin, H.J.; Lee, Y.; Joo, K.M.; Lee, J.; Nam, D.H. Wnt/beta-catenin signaling is a key downstream mediator of met signaling in glioblastoma stem cells. Neuro. Oncol. 2013, 15, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-MET pathway potentiates glioblastoma responses to gamma-radiation. Clin. Cancer Res. 2005, 11, 4479–4486. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.W.; Mahadevan, D.; Ellsworth, R.; Cooke, L.; Bearss, D.; Stea, B. The c-MET receptor tyrosine kinase inhibitor mp470 radiosensitizes glioblastoma cells. Radiat. Oncol. 2009, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, I.M.; Scott, T.; Tandle, A.T.; Burgan, W.E.; Burgess, T.L.; Tofilon, P.J.; Camphausen, K. Radiosensitization of glioma cells by modulation of MET signalling with the hepatocyte growth factor neutralizing antibody, AMG102. J. Cell Mol. Med. 2011, 15, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.T.; Sun, J.; Rex, K.; Radinsky, R.; Kendall, R.; Coxon, A.; Burgess, T.L. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in u-87 MG cells and xenografts. Clin. Cancer Res. 2007, 13, 6735–6742. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Narayan, R.N.; Horton, L.; Patel, T.R.; Habib, A.A. The role of EGFR-MET interactions in the pathogenesis of glioblastoma and resistance to treatment. Curr. Cancer Drug Targets 2016, 17, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Acquaviva, J.; Chi, D.; Lessard, J.; Zhu, H.; Woolfenden, S.; Bronson, R.T.; Pfannl, R.; White, F.; Housman, D.E.; et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2012, 31, 3039–3050. [Google Scholar] [CrossRef] [PubMed]
- Nehoff, H.; Parayath, N.N.; McConnell, M.J.; Taurin, S.; Greish, K. A combination of tyrosine kinase inhibitors, crizotinib and dasatinib for the treatment of glioblastoma multiforme. Oncotarget 2015, 6, 37948–37964. [Google Scholar] [PubMed]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Wullich, B.; Sattler, H.P.; Fischer, U.; Meese, E. Two independent amplification events on chromosome 7 in glioma: Amplification of the epidermal growth factor receptor gene and amplification of the oncogene MET. Anticancer Res. 1994, 14, 577–579. [Google Scholar] [PubMed]
- Jun, H.J.; Bronson, R.T.; Charest, A. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells 2014, 32, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Liffers, K.; Kathagen, A.; Riethdorf, S.; Zapf, S.; Merlo, A.; Kolbe, K.; Westphal, M.; Lamszus, K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRVIII and PI3Kp110delta. Neuro. Oncol. 2013, 15, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Garnett, J.; Chumbalkar, V.; Vaillant, B.; Gururaj, A.E.; Hill, K.S.; Latha, K.; Yao, J.; Priebe, W.; Colman, H.; Elferink, L.A.; et al. Regulation of hgf expression by deltaEGFR-mediated c-MET activation in glioblastoma cells. Neoplasia 2013, 15, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.; Kim, S.I.; Park, C.K.; Paek, S.H.; Lee, S.T.; Park, S.H. c-MET overexpression and amplification in gliomas. Int. J. Clin. Exp. Pathol. 2015, 8, 14932–14938. [Google Scholar] [PubMed]
- Cui, J.J. Targeting receptor tyrosine kinase met in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-MET signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Nakamura, T. HGF-MET cascade, a key target for inhibiting cancer metastasis: The impact of NK4 discovery on cancer biology and therapeutics. Int. J. Mol. Sci. 2013, 14, 888–919. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.E.; Rex, K.; Caenepeel, S.; Yang, Y.; Zhang, Y.; Broome, M.A.; Kha, H.T.; Burgess, T.L.; Amore, B.; Kaplan-Lefko, P.J.; et al. In vitro and in vivo activity of AMG 337, a potent and selective MET kinase inhibitor, in MET-dependent cancer models. Mol. Cancer Ther. 2016, 15, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Berthou, S.; Aebersold, D.M.; Schmidt, L.S.; Stroka, D.; Heigl, C.; Streit, B.; Stalder, D.; Gruber, G.; Liang, C.; Howlett, A.R.; et al. The MET kinase inhibitor su11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene 2004, 23, 5387–5393. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.H.; Liu, L.; Lee, M.; Xi, N.; Fellows, I.; D’Angelo, N.D.; Dominguez, C.; Rex, K.; Bellon, S.F.; Kim, T.S.; et al. Structure-based design of novel class II c-MET inhibitors: 1. Identification of pyrazolone-based derivatives. J. Med. Chem. 2012, 55, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Norman, M.H.; Lee, M.; Xi, N.; Siegmund, A.; Boezio, A.A.; Booker, S.; Choquette, D.; D’Angelo, N.D.; Germain, J.; et al. Structure-based design of novel class II c-MET inhibitors: 2. Sar and kinase selectivity profiles of the pyrazolone series. J. Med. Chem. 2012, 55, 1868–1897. [Google Scholar] [CrossRef] [PubMed]
- Tiedt, R.; Degenkolbe, E.; Furet, P.; Appleton, B.A.; Wagner, S.; Schoepfer, J.; Buck, E.; Ruddy, D.A.; Monahan, J.E.; Jones, M.D.; et al. A drug resistance screen using a selective met inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011, 71, 5255–5264. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.M.; et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014, 74, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Leonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 is a novel kinase inhibitor of met, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRVIII in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Shapiro, G.I. Crizotinib, a small-molecule dual inhibitor of the c-MET and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs 2010, 11, 1477–1490. [Google Scholar] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guessous, F.; Kofman, A.; Schiff, D.; Abounader, R. Xl-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 2010, 13, 112–121. [Google Scholar] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Schuler, M.; Berardi, R.; Lim, W.T.; Van Geel, R.; De Jonge, M.; Azaro, A.; Gottfried, M.; Han, J.Y.; Lee, D.H.; et al. Mini01.03: Phase (ph) I study of the safety and efficacy of the c-MET inhibitor capmatinib (INC280) in patients with advanced CMET+ NSCLC: Topic: Medical oncology. J. Thorac. Oncol. 2016, 11, S257–S258. [Google Scholar] [CrossRef] [PubMed]
- Friese-Hamim, M.; Bladt, F.; Locatelli, G.; Stammberger, U.; Blaukat, A. The selective c-MET inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c-MET activation in NSCLC models. Am. J. Cancer Res. 2017, 7, 962–972. [Google Scholar] [PubMed]
- Shin, T.H.; Sung, E.S.; Kim, Y.J.; Kim, K.S.; Kim, S.H.; Kim, S.K.; Lee, Y.D.; Kim, Y.S. Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting peptide improves the antitumor efficacy. Mol. Cancer Ther. 2014, 13, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Chekhonin, I.V.; Leopold, A.V.; Gurina, O.I.; Semenova, A.V. Monoclonal antibodies in high-grade gliomas. Vestn. Akad. Med. Nauk SSSR 2014, 9–10, 131–139. [Google Scholar] [CrossRef]
- Sharma, N.; Adjei, A.A. In the clinic: Ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther. Adv. Med. Oncol. 2011, 3, S37–S50. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Joo, K.M.; Lee, J.; Yoon, Y.; Nam, D.H. Targeting the epithelial to mesenchymal transition in glioblastoma: The emerging role of met signaling. Onco.Targets Ther. 2014, 7, 1933–1944. [Google Scholar] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Kouri, F.M.; Ritner, C.; Stegh, A.H. miRNA-182 and the regulation of the glioblastoma phenotype—Toward miRNA-based precision therapeutics. Cell Cycle 2015, 14, 3794–3800. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.A.; Zhang, Y.; Curley, C.; Price, R.L.; Abounader, R. 370 magnetic resonance-guided focused ultrasound delivery of polymeric brain-penetrating nanoparticle microRNA conjugates in glioblastoma. Neurosurgery 2016, 63, 210. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Finniss, S.; Cazacu, S.; Bucris, E.; Ziv-Av, A.; Xiang, C.; Bobbitt, K.; Rempel, S.A.; Hasselbach, L.; Mikkelsen, T.; et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget 2013, 4, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Doucette, T.; Rao, G.; Yang, Y.; Gumin, J.; Shinojima, N.; Bekele, B.N.; Qiao, W.; Zhang, W.; Lang, F.F. Mesenchymal stem cells display tumor-specific tropism in an RCAS/NTV—A glioma model. Neoplasia 2011, 13, 716–725. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Phase | Status | Drug | Combinations | Patient Population | Results |
|---|---|---|---|---|---|---|
| NCT 02386826 | Ib | Recruiting | INCB28060 (INC280) | Bevacizumab | Recurrent GBM, Metastatic Colorectal Cancer (mCRC) Metastatic Renal Cell Carcinoma (mRCC) | None Reported |
| NCT 01870726 | II | Completed | INCB28060 (INC280) | Buparlisib | Recurrent GBM | None reported |
| NCT 01441388 | Ib | Withdrawn | Crizotinib | VEGF inhibitors, axitinib, sunitinib, bevacizumab and sorafenib | Advanced solid tumors in GBM, Renal Cell Carcinoma (RCC) and Hepatocellular Carcinoma (HCC) | None Reported |
| NCT 00939770 | I/II | Active, not recruiting | Crizotinib | - | Relapsed/refractory solid tumors in brain and central nervous system, neuroblastoma and anaplastic large cell lymphoma | None Reported |
| NCT 01644773 | I | Recruiting | Crizotinib | Dasatinib | High-grade glioma, diffuse intrinsic pontine glioma | None Reported |
| NCT 02034981 | II | Recruiting | Crizotinib | - | MET amplified GBM | None Reported |
| NCT 01632228 | II | Completed | Onartuzumab | Onartuzumab with bevacizumab versus bevacizumab alone or onartuzumab monotherapy | Recurrent GBM | None reported |
| NCT 01113398 | II | Completed | Rilotumumab | Bevacizumab | Recurrent malignant glioma | 16.67% of cohort experienced serious adverse events |
| NCT 00427440 | II | Completed | Rilotumumab | - | Advanced malignant glioma | None reported |
| NCT 00960492 | I | Completed | Cabozantinib | Temozolomide, radiation therapy | GBM, Giant Cell GBM and Gliosarcoma | None reported |
| NCT 00704288 | II | Completed | Cabozantinib | - | Recurrent GBM | None reported |
| NCT 01189513 | I | Withdrawn | Ficlatuzumab | - | GBM | None Reported |
| NCT 01433991 | I/II | Active, not recruiting | Golvatinib | Lenvatinib | Recurrent GBM, unresectable stage III/IV melanoma | None Reported |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. https://doi.org/10.3390/cancers9070087
Cruickshanks N, Zhang Y, Yuan F, Pahuski M, Gibert M, Abounader R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers. 2017; 9(7):87. https://doi.org/10.3390/cancers9070087
Chicago/Turabian StyleCruickshanks, Nichola, Ying Zhang, Fang Yuan, Mary Pahuski, Myron Gibert, and Roger Abounader. 2017. "Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma" Cancers 9, no. 7: 87. https://doi.org/10.3390/cancers9070087
APA StyleCruickshanks, N., Zhang, Y., Yuan, F., Pahuski, M., Gibert, M., & Abounader, R. (2017). Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers, 9(7), 87. https://doi.org/10.3390/cancers9070087