Solvent Influence on Selectivity in α-Pinene Oxide Isomerization Using MoO3-Modified Zeolite BETA


Abstract
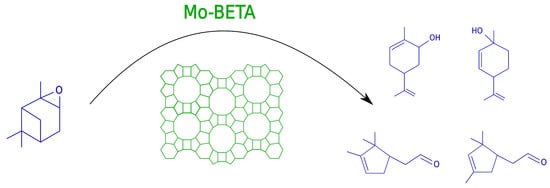
:
1. Introduction
2. Results and Discussion
2.1. Material Preparation and Characterization
2.2. Catalytic Testing
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Edl, W.; Sienel, G.R. Process for the production of epoxides. U.S. Patent No. 4882442A, 30 December 1989. [Google Scholar]
- Jaafari, A.; Tilaoui, M.; Mouse, H.A.; M’bark, L.A.; Aboufatima, R.; Chait, A.; Lepoivre, M.; Zyad, A. Comparative study of the antitumor effect of natural monoterpenes: Relationship to cell cycle analysis. Revista Brasileira Farmacognosia 2012, 22, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.I.; Anwar, F.; Shahid, M.; Ashraf, M.; Przybylski, R. Chemical Composition, and Antioxidant and Antimicrobial Activities of Essential Oil of Spearmint (Mentha spicata L.). J. Essent. Oil Res. 2010, 22, 78–84. [Google Scholar] [CrossRef]
- Castro, J.M.; Linares-Palomino, P.J.; Salido, S.; Altarejos, J.; Nogueras, M.; Sánchez, A. Enantiospecific synthesis, separation and olfactory evaluation of all diastereomers of a homologue of the sandalwood odorant Polysantol. Tetrahedron 2005, 61, 11192–11203. [Google Scholar] [CrossRef]
- Arbushow, B. Studium der Isomerisation von Terpen-oxyden, I. Mitteil.: Isomerisation des α-Pinen-oxydes bei der Reaktion von Reformatsky. Chem. Ber. 1935, 68, 1430–1435. [Google Scholar] [CrossRef]
- Duetz, W.; Fjällman, A.; Ren, S.; Jourdat, C.; Witholt, B. Biotransformation of D-Limonene to (+) trans-Carveol by Toluene-Grown Rhodococcus opacus PWD4 Cells. Appl. Environ. Microbiol. 2001, 67, 2829–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, J.V.; de Meireles, A.L.P.; da Silva Rocha, K.A.; Pereira, M.C.; Oliveira, L.C.A.; Gusevskaya, E.V. Isomerization of α-pinene oxide catalyzed by iron-modified mesoporous silicates. Appl. Catal. A Gen. 2012, 443–444, 125–132. [Google Scholar] [CrossRef]
- Kumar, N.; Mäki-Arvela, P.; Diaz, S.F.; Aho, A.; Demidova, Y.; Linden, J.; Shepidchenko, A.; Tenhu, M.; Salonen, J.; Laukkanen, P.; et al. Isomerization of α-Pinene Oxide Over Iron-Modified Zeolites. Top. Catal. 2013, 56, 696–713. [Google Scholar] [CrossRef]
- Ravasio, N.; Zaccheria, F.; Gervasini, A.; Messi, C. A new, Fe based, heterogeneous Lewis acid: Selective isomerization of α-pinene oxide. Catal. Commun. 2008, 9, 1125–1127. [Google Scholar] [CrossRef]
- Štekrová, M.; Kumar, N.; Aho, A.; Sinev, I.; Grunert, W.; Dahl, J.; Roine, J.; Arzumanov, S.S.; Mäki-Arvela, P.; Murzin, D.Y. Isomerization of alpha-pinene oxide using Fe-supported catalysts: Selective synthesis of campholenic aldehyde. Appl. Catal. A Gen. 2014, 470, 162–176. [Google Scholar] [CrossRef]
- Sánchez-Velandia, J.E.; Villa, A.L. Isomerization of α- and β-pinene epoxides over Fe or Cu supported MCM-41 and SBA-15 materials. Appl. Catal. A Gen. 2019, 580, 17–27. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Alvaro, M.; Chevreau, H.; Horcajada, P.; Devic, T.; Serre, C.; Garcia, H. Iron(iii) metal–organic frameworks as solid Lewis acids for the isomerization of α-pinene oxide. Catal. Sci. Technol. 2012, 2, 324–330. [Google Scholar] [CrossRef]
- Fellenz, N.A.; Bengoa, J.F.; Marchetti, S.G.; Gervasini, A. Influence of the Brönsted and Lewis acid sites on the catalytic activity and selectivity of Fe/MCM-41 system. Appl. Catal. A Gen. 2012, 435-436, 187–196. [Google Scholar] [CrossRef]
- Vyskočilová, E.; Hašková, L.; Červený, L. Solvent-induced selectivity in α-pinene oxide isomerization catalyzed by Fe-modified zeolite beta. Chem. Pap. 2019, 73, 1621–1627. [Google Scholar] [CrossRef]
- Štekrová, M.; Kumar, N.; Diaz, S.F.; Mäki-Arvela, P.; Murzin, D.Y. H- and Fe-modified zeolite beta catalysts for preparation of trans-carveol from α-pinene oxide. Catal. Today 2015, 241, 237–245. [Google Scholar] [CrossRef]
- Kunkeler, P.J.; van der Waal, J.C.; Bremmer, J.; Zuurdeeg, B.J.; Downing, R.S.; van Bekkum, H. Application of zeolite titanium Beta in the rearrangement of α-pinene oxide to campholenic aldehyde. Catal. Lett. 1998, 53, 135–138. [Google Scholar] [CrossRef]
- Panadero, M.P.; Velty, A. Readily available Ti-beta as an efficient catalyst for greener and sustainable production of campholenic aldehyde. Catal. Sci. Technol. 2019, 9, 4293–4303. [Google Scholar] [CrossRef]
- Pitínová-Štekrová, M.; Eliášová, P.; Weissenberger, T.; Shamzy, M.; Musilová, Z.; Čejka, J. Highly selective synthesis of campholenic aldehyde over Ti-MWW catalysts by α-pinene oxide isomerization. Catal. Sci. Technol. 2018, 8, 4690–4701. [Google Scholar] [CrossRef]
- Costa, V.V.; da Silva Rocha, K.A.; de Sousa, L.F.; Robles-Dutenhefner, P.A.; Gusevskaya, E.V. Isomerization of α-pinene oxide over cerium and tin catalysts: Selective synthesis of trans-carveol and transsobrerol. J. Mol. Catal. A Chem. 2011, 345, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Štekrová, M.; Matoušková, M.; Vyskočilová, E.; Červený, L. Selective preparation of campholenic aldehyde over heterogenized methyltrioxorhenium. Res. Chem. Intermed. 2015, 41, 9003–9013. [Google Scholar] [CrossRef]
- Štekrová, M.; Kumar, N.; Mäki-Arvela, P.; Ardashov, O.V.; Volcho, K.P.; Salakhutdinov, N.F.; Murzin, D.Y. Selective Preparation of trans-Carveol over Ceria Supported Mesoporous Materials MCM-41 and SBA-15. Materials 2013, 6, 2103–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Rocha, K.A.; Kozhevnikov, I.V.; Gusevskaya, E.V. Isomerisation of α-pinene oxide over silica supported heteropoly acid H3PW12O40. Appl. Catal. A 2005, 294, 106–110. [Google Scholar] [CrossRef]
- Da Silva Rocha, K.A.; Hoehne, J.L.; Gusevskaya, E.V. Phosphotungstic Acid as a Versatile Catalyst for the Synthesis of Fragrance Compounds by α-Pinene Oxide Isomerization: Solvent-Induced Chemoselectivity. Chem. Eur. J. 2008, 14, 6166–6172. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, A.Y.; Ignatovich, Z.V.; Ermolinskaya, A.L.; Kravtsova, A.V.; Baranovskii, A.V.; Koroleva, E.V.; Agabekov, V.E. Synthesis of Fencholenic Aldehyde from α-pinene Epoxide on Modified Clays. Chem. Nat. Compd. 2018, 54, 893–897. [Google Scholar] [CrossRef]
- Vrbková, E.; Šteflová, B.; Sekerová, L.; Krupka, J.; Vyskočilová, E.; Červený, L. Contribution to MoO 3–SiO 2 and WO 3–SiO 2 utilization—Active catalysts in jasmine aldehyde, 2-hexyl-1, 3-dioxolane and methyllaurate synthesis. React. Kinet. Mech. Catal. 2020, 129, 645–658. [Google Scholar] [CrossRef]
- Vrbková, E.; Šteflová, B.; Vyskočilová, E.; Červený, L. Heterogeneous Mo/W/Zn–SiO2 based catalysts in nopol (2-(6,6-dimethyl-2-bicyclo[3.1.1]hept-2-enyl)ethanol) synthesis. React. Kinet. Mech. Catal. 2020, 131, 213–232. [Google Scholar] [CrossRef]
- Wehrer, W.; Bigey, L.; Hilaire, L. Catalytic reactions of n-hexane and 1-hexene on molybdenum dioxide. Appl. Catal. A 2003, 243, 109–119. [Google Scholar] [CrossRef]
- Bouchy, C.; Pham-Huu, C.; Heinrich, B.; Chaumont, C.; Ledoux, M.J. Microstructure and Characterization of a Highly Selective Catalyst for the Isomerization of Alkanes: A Molybdenum Oxycarbide. J. Catal. 2000, 190, 92–103. [Google Scholar] [CrossRef]
- Lamic, A.F.; Shin, C.H.; Djega-Mariadassou, G.; Potvin, C. Characterization of Mo2C–WO2 composite catalysts for bifunctional isomerization: A new pulse method to quantify acid sites. Appl. Catal. A 2006, 302, 5–13. [Google Scholar] [CrossRef]
- York, A.P.E.; Pham-Huu, C.; Del Gallo, P.; Ledoux, M.J. Molybdenum oxycarbide hydrocarbon isomerization catalysts: Cleaner fuels for the future. Catal. Today 1997, 35, 51–57. [Google Scholar] [CrossRef]
- Ohno, T.; Li, Z.; Sakai, N.; Sakagami, H.; Takahashi, N.; Matsuda, T. Heptane isomerization over molybdenum oxides obtained by H2 reduction of HxMoO3 with different hydrogen contents. Appl. Catal. A 2010, 389, 52–59. [Google Scholar] [CrossRef]
- Bruno, S.M.; Valente, A.A.; Pillinger, M.; Amelse, J.; Romão, C.C.; Gonçalves, I.S. Efficient Isomerization of α-Pinene Oxide to Campholenic Aldehyde Promoted by a Mixed-Ring Analogue of Molybdenocene. ACS Sustain. Chem. Eng. 2019, 7, 13639–13645. [Google Scholar] [CrossRef]
- Caullet, P.; Hazm, J.; Guth, J.; Joly, J.; Lynch, J.; Raatz, F. Synthesis of zeolite Beta from nonalkaline fluoride aqueous aluminosilicate gels. Zeolites 1992, 12, 240–250. [Google Scholar] [CrossRef]
- Hansen, S.; Andersson, A. Electron microscopy of some molybdenum oxide phases after use as catalysts in oxidative ammonolysis and ammoxidation of toluene. J. Solid State Chem. 1988, 75, 225–243. [Google Scholar] [CrossRef]
- Sekerová, L.; Vyskočilová, E.; Červený, L. Prins cyclization of isoprenol with various aldehydes using MoO3/SiO2 as a catalyst. React. Kinet. Mech. Catal. 2017, 121, 83–95. [Google Scholar] [CrossRef]
- Gutmann Acceptor and Donor number. Available online: http://www.stenutz.eu/chem/solv21.php (accessed on 12 August 2020).
- Solvent Properties Chart. Available online: https://depts.washington.edu/eooptic/linkfiles/dielectric_chart%5B1%5D.pdf (accessed on 12 August 2020).
- Munack, A.; Schmidt, L.; Schröder, O.; Schaper, K.; Pabst, C.; Krahl, J. Alcohols as a means to inhibit the formation of precipitates in blends of biodiesel and fossil diesel fuel. Agric. Eng. Int. 2015, 2015, 226–233. [Google Scholar]
- Dielectric Constants of Liquids. Available online: https://www.engineeringtoolbox.com/liquid-dielectric-constants-d_1263.html (accessed on 31 August 2020).
- Dielectric Constants of Common Materials. Available online: https://www.kabusa.com/Dilectric-Constants.pdf (accessed on 31 August 2020).
- Mäki-Arvela, P.; Shcherban, N.; Lozachmeur, C.; Russo, V.; Wärnå, J.; Murzin, D.Y. Isomerization of α-Pinene Oxide: Solvent Effects, Kinetics and Thermodynamics. Catal. Lett. 2019, 149, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Vrbková, E.; Šteflová, B.; Zapletal, M.; Vyskočilová, E.; Červený, L. Tungsten oxide-based materials as effective catalysts in isopulegol formation by intramolecular Prins reaction of citronellal. Res. Chem. Intermed. 2020, 46, 4047–4059. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | MoO3 (%) | Mo (%) calculated | SiO2 (%) | Al2O3 (%) | Others (%) |
|---|---|---|---|---|---|
| 20Mo450 | 32.8 | 21.6 | 63.7 | 3.4 | 0.1 |
| 20Mo450RT | 36.0 | 23.7 | 60.8 | 3.1 | 0.1 |
| 20Mo500 | 32.0 | 21.1 | 64.7 | 3.2 | 0.1 |
| 20Mo550 | 33.2 | 21.9 | 63.5 | 3.2 | 0.1 |
| 20Mo600 | 33.9 | 22.3 | 62.9 | 3.1 | 0.1 |
| Material | SBET m2/g | St-plot m2/g | Total Pore Volume cm3/g | t-Plot Micropore Volume cm3/g | Ratio of Micropores (%) |
|---|---|---|---|---|---|
| BETA38 | 539.62 | 136.15 | 0.341 | 0.210 | 61.6 |
| 20Mo450 | 272.81 | 58.70 | 0.181 | 0.110 | 60.8 |
| 20Mo500 | 239.46 | 53.98 | 0.171 | 0.096 | 56.1 |
| 20Mo550 | 154.33 | 43.47 | 0.139 | 0.057 | 41.0 |
| 20Mo600 | 16.50 | 12.43 | 0.057 | 0.002 | 3.5 |
| 20Mo450RT | 269.30 | 61.61 | 0.184 | 0.107 | 58.1 |
| Material | Span Value | Particle Size (µm) | ||
|---|---|---|---|---|
| Dv(10) | Dv(50) | Dv(90) | ||
| BETA38 | 1.6 | 0.39 | 0.71 | 1.6 |
| 20Mo450 | 22.2 | 0.43 | 1.64 | 35.2 |
| 20Mo450RT | 22.5 | 0.44 | 1.94 | 44.2 |
| 20Mo500 | 47.9 | 0.46 | 1.74 | 55.0 |
| 20Mo550 | 81.9 | 0.50 | 4.74 | 389.0 |
| 20Mo600 | 43.8 | 0.58 | 16.20 | 710.0 |
| Solvent | Solvent Type | Donor Number (kJ/mol) | Dielectric Constant (Relative Permitivity) | Initial Reaction Rate (mmol/gcat.min) | APO Conversion at 3 h (%) | CA Selectivity at 3 h (%) | TCA Selectivity at 3 h (%) | PMD Selectivity at 3 h (%) |
|---|---|---|---|---|---|---|---|---|
| cyclohexane | nonpolar | 0 | 2.02 | 2.40 | 99 | 34.6 | 17.4 | 13.9 |
| toluene | nonpolar | 0.1 | 2.38 | 5.43 | 100 | 34.2 | 14.8 | 14.0 |
| 1,4 dioxane | nonpolar | 14.3 | 2.25 | 6.63 | 100 | 28.7 | 18.0 | 11.0 |
| nitromethane | polar aprotic | 2.7 | 35.87 | 4.68 | 86 | 44.9 | 10.7 | 12.3 |
| butan-1-ol | polar protic | 19.5 | 17.8 | 13.11 | 100 | 3.2 | 7.0 | 1.9 |
| propan-1-ol | polar protic | 19.8 | 21.8 | 13.11 | 100 | 3.3 | 6.4 | 2.7 |
| propan-2-ol | polar protic | 21.1 | 17.9 | 13.11 | 100 | 18.2 | 14.2 | 5.4 |
| dichlorbenzene | polar aprotic | 3 | 9.93 | 2.67 | 100 | 42.1 | 13.3 | 13.1 |
| benzonitrile | polar aprotic | 11.9 | 26 | 0.71 | 16 | 66.5 | 9.8 | 0.0 |
| acetonitrile | polar aprotic | 14 | 37.5 | 0.68 | 10 | 62.7 | 4.9 | 2.9 |
| ethylacetate | polar aprotic | 17.1 | 6.02 | 2.67 | 97 | 37.1 | 15.8 | 13.9 |
| pentan-2-one | polar aprotic | 17.1 | 18.2 | 11.32 | 100 | 35.1 | 15.5 | 12.0 |
| butan-2-one | polar aprotic | 17.4 | 18.5 | 11.95 | 100 | 30.8 | 10.4 | 9.1 |
| cyclohexanone | polar aprotic | 18 | 18.2 | 8.34 | 100 | 37.4 | 15.0 | 16.6 |
| tetrahydrofuran | polar aprotic | 20 | 7.58 | 6.74 | 100 | 31.7 | 28.9 | 6.5 |
| cyclohexanol | polar protic | 25 | 15 | 5.58 | 100 | 24.1 | 21.1 | 5.1 |
| N,N’-dimethylformamide | polar aprotic | 26.6 | 36.7 | 1.01 | 27 | 26.5 | 43.8 | 14.0 |
| N-methylpyrrolidone | polar aprotic | 27.3 | 32.17 | 0.85 | 41 | 26.1 | 42.9 | 12.2 |
| N,N’-dimethylacetamide | polar aprotic | 27.8 | 37.8 | 4.65 | 44 | 25.3 | 45.5 | 11.5 |
| dimethylsulfoxide | polar aprotic | 29.8 | 46.7 | 0.97 | 45 | 25.3 | 53.6 | 12.7 |
| pyridine | polar aprotic | 33.1 | 12.4 | 0.14 | 3 | 55.6 | 18.5 | 0.0 |
| Material Denotation | Temperature of Wet Impregnation | Calcination Temperature (°C) |
|---|---|---|
| 20Mo450RT | room temperature | 450 |
| 20Mo450 | 60 °C | 450 |
| 20Mo500 | 500 | |
| 520Mo550 | 550 | |
| 20Mo600 | 600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrbková, E.; Vyskočilová, E.; Lhotka, M.; Červený, L. Solvent Influence on Selectivity in α-Pinene Oxide Isomerization Using MoO3-Modified Zeolite BETA. Catalysts 2020, 10, 1244. https://doi.org/10.3390/catal10111244
Vrbková E, Vyskočilová E, Lhotka M, Červený L. Solvent Influence on Selectivity in α-Pinene Oxide Isomerization Using MoO3-Modified Zeolite BETA. Catalysts. 2020; 10(11):1244. https://doi.org/10.3390/catal10111244
Chicago/Turabian StyleVrbková, Eva, Eliška Vyskočilová, Miloslav Lhotka, and Libor Červený. 2020. "Solvent Influence on Selectivity in α-Pinene Oxide Isomerization Using MoO3-Modified Zeolite BETA" Catalysts 10, no. 11: 1244. https://doi.org/10.3390/catal10111244
APA StyleVrbková, E., Vyskočilová, E., Lhotka, M., & Červený, L. (2020). Solvent Influence on Selectivity in α-Pinene Oxide Isomerization Using MoO3-Modified Zeolite BETA. Catalysts, 10(11), 1244. https://doi.org/10.3390/catal10111244