Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT

 ,
, 
Abstract
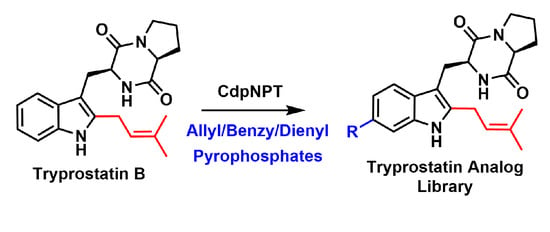
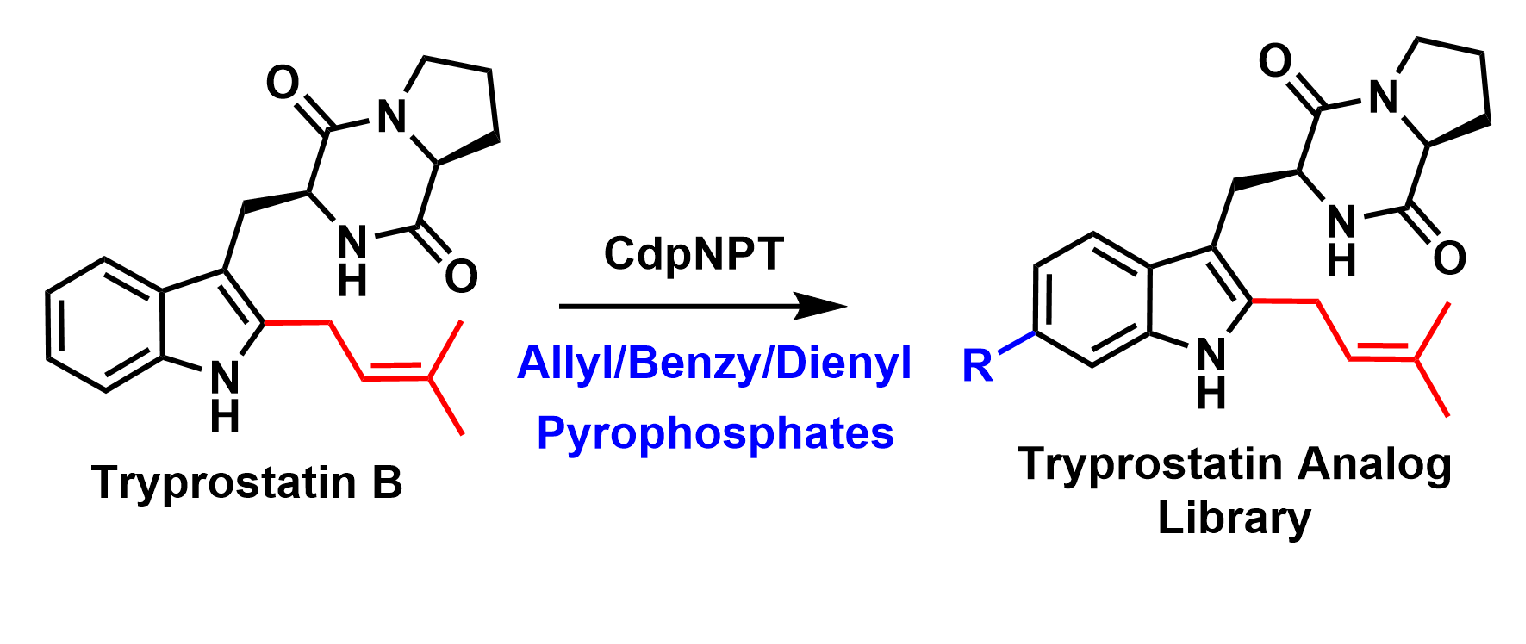
:
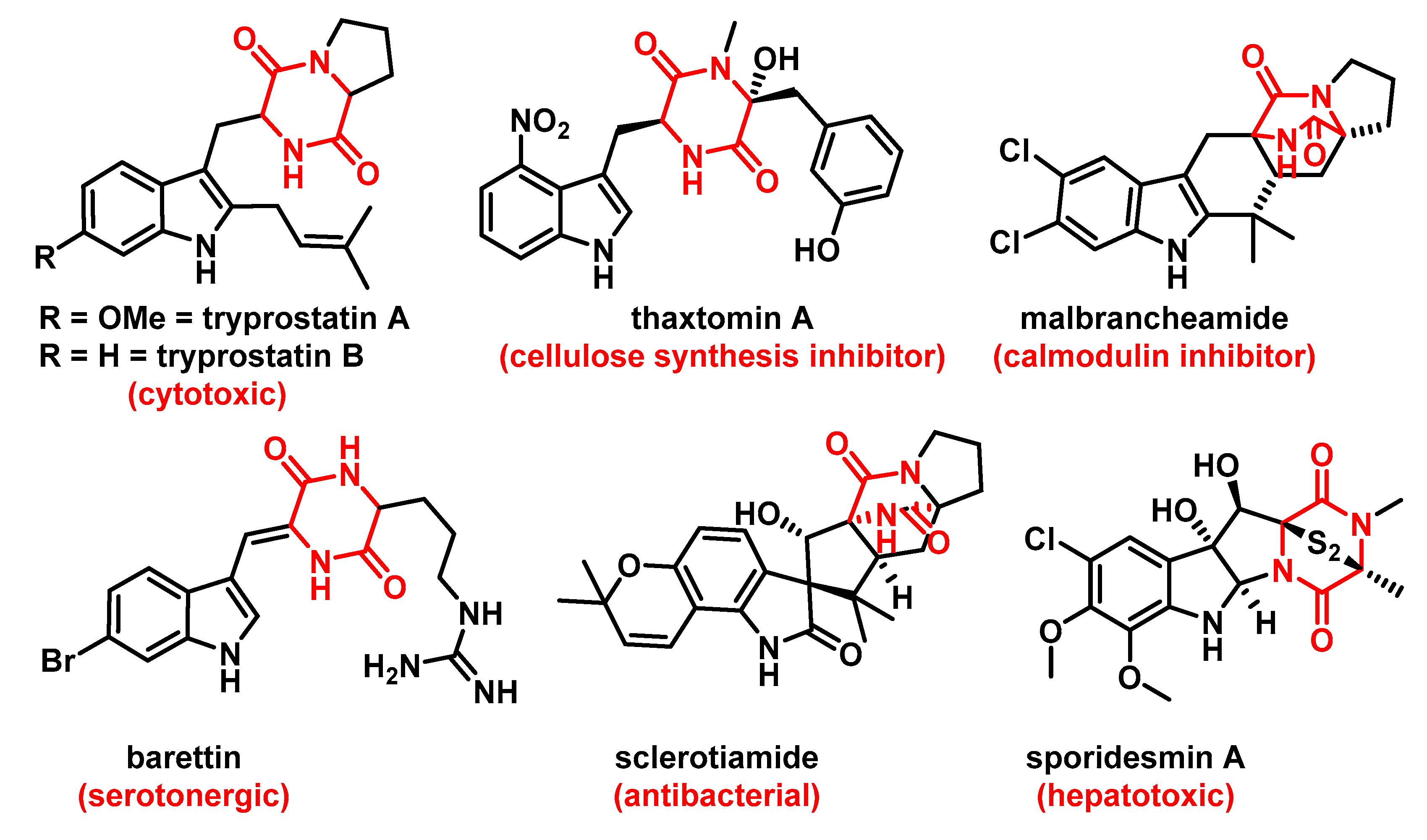
1. Introduction
2. Results and Discussion
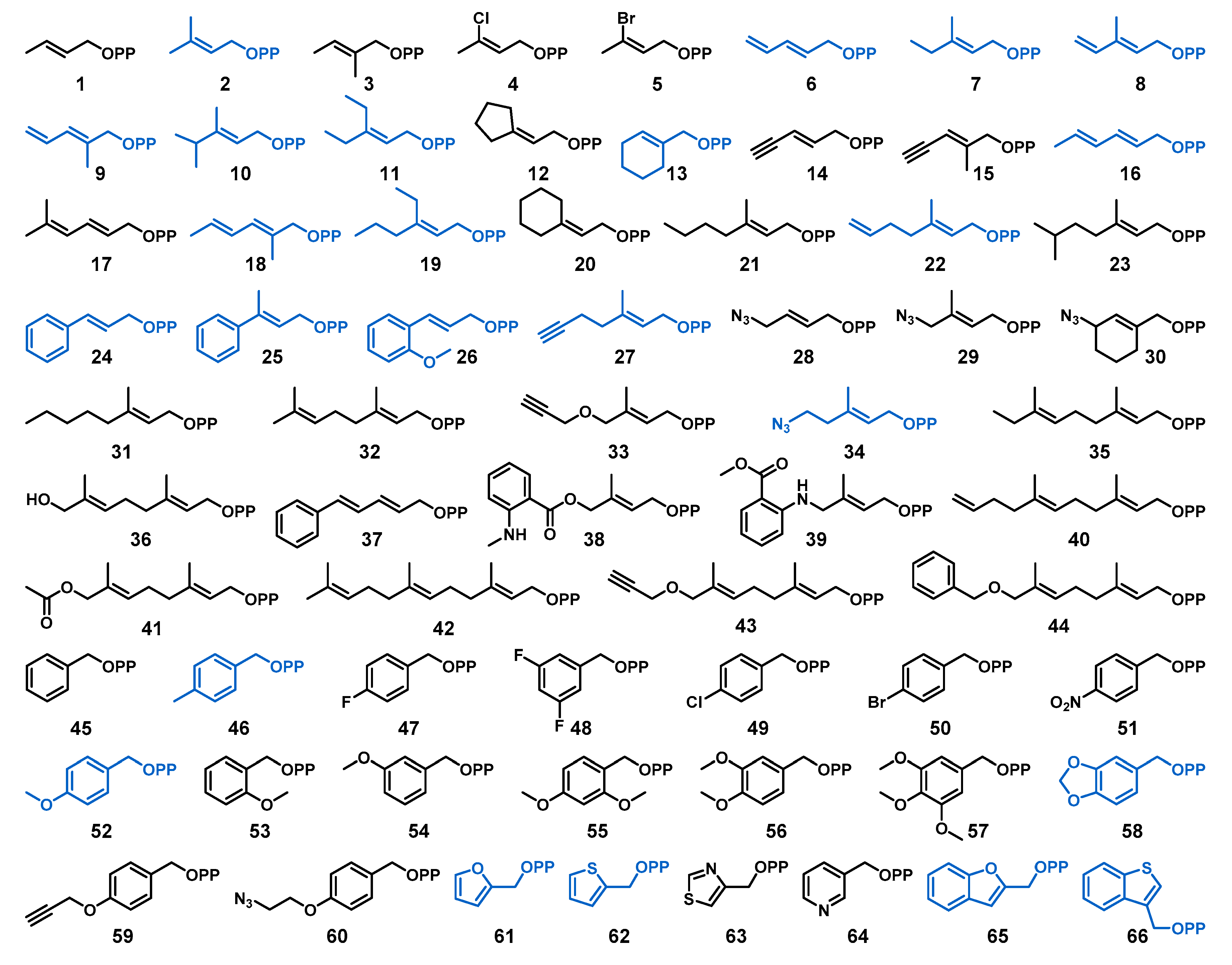
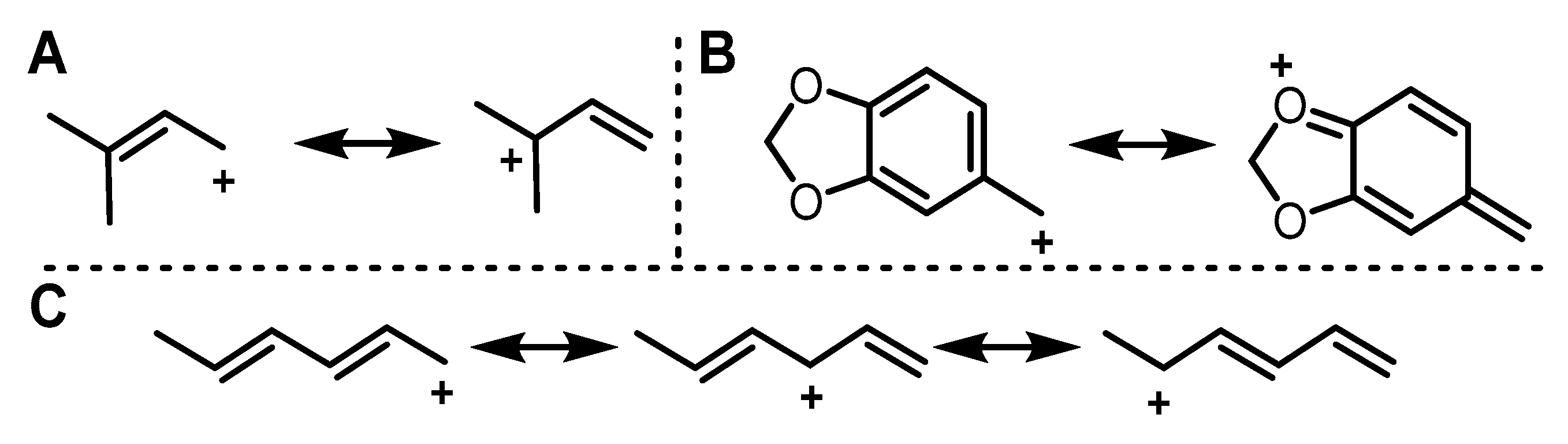
2.1. Design of Alkyl Pyrophosphate Donors
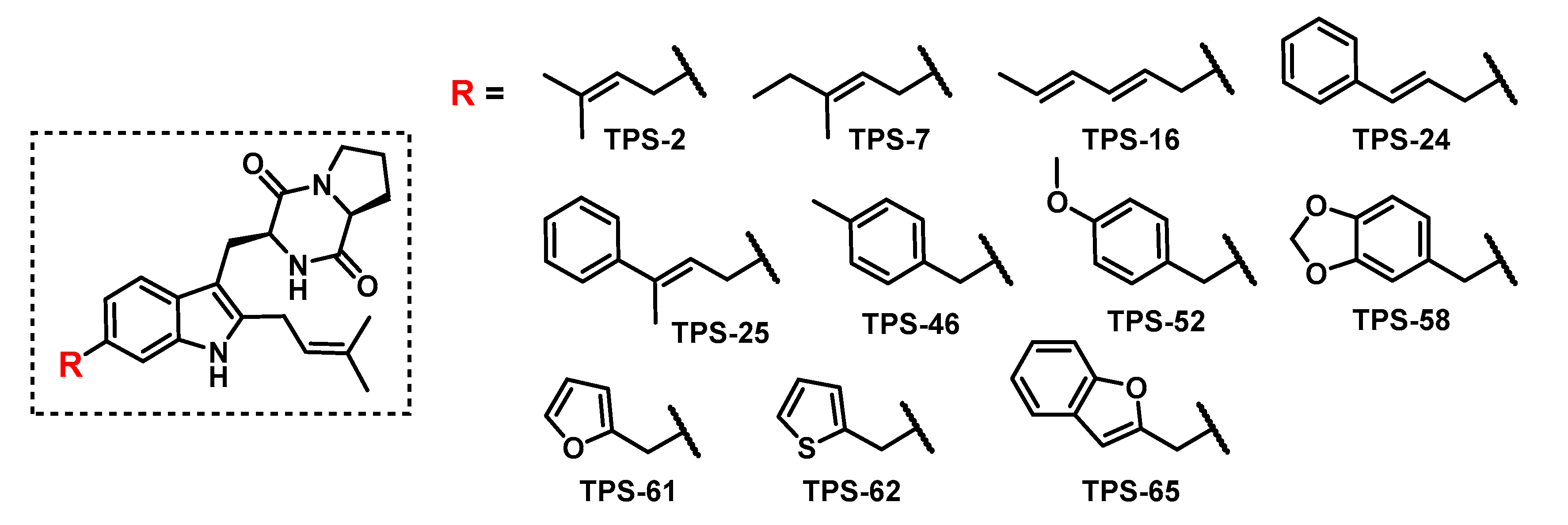
2.2. Donor Library Screening
2.3. Allylic Donors
2.4. Benzylic and Heterocyclic Donors
2.5. Other Functionalized Donors
2.6. Scale-Up and Characterization of CdpNPT Reaction Products
2.7. Cytotoxicity Studies
3. Materials and Methods
3.1. General Materials
3.2. General Methods
3.3. In Vitro CdpNPT Assay
3.4. Enzymatic Scale-Up Reactions
3.5. Determination of Structures
3.6. HPLC Method A
3.7. HPLC Method B
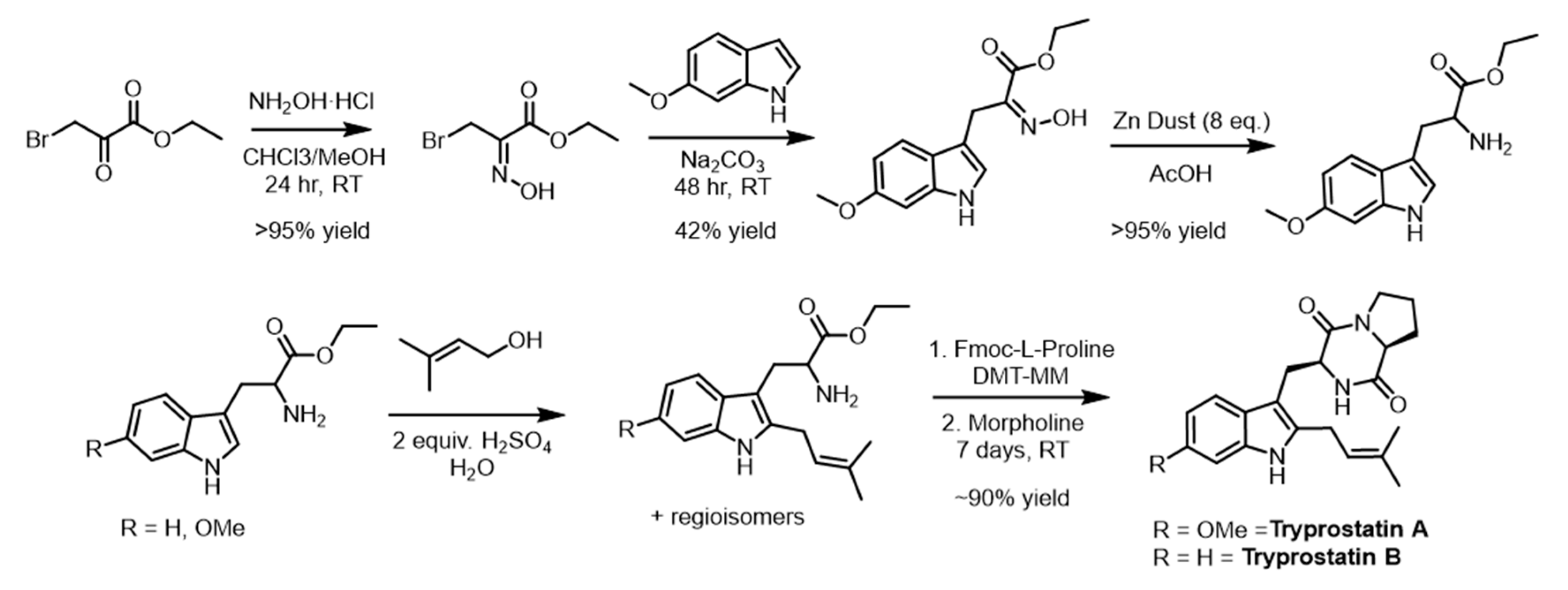
3.8. Synthesis Tryprostatin A and B
3.9. Cell Titer-Blue Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evidente, A.; Kornienko, A.; Cimmino, A.; Andolfi, A.; Lefranc, F.; Mathieu, V.; Kiss, R. Fungal metabolites with anticancer activity. Nat. Prod. Rep. 2014, 31, 617–627. [Google Scholar] [CrossRef]
- Ma, Y.-M.; Liang, X.-A.; Kong, Y.; Jia, B. Structural Diversity and Biological Activities of Indole Diketopiperazine Alkaloids from Fungi. J. Agric. Food Chem. 2016, 64, 6659–6671. [Google Scholar] [CrossRef]
- Cui, C.-B.; Kakeya, H.; Okada, G.; Onose, R.; Osada, H. Novel Mammalian Cell Cycle Inhibitors, Tryprostatins A, B and Other Diketopiperazines Produced by Aspergillus fumigatus. I. Taxonomy, Fermentation, Isolation and Biological Properties. J. Antibiot. 1996, 49, 527–533. [Google Scholar] [CrossRef]
- Bischoff, V.; Cookson, S.J.; Wu, S.; Scheible, W.-R. Thaxtomin A affects CESA-complex density, expression of cell wall genes, cell wall composition, and causes ectopic lignification in Arabidopsis thaliana seedlings. J. Exp. Bot. 2009, 60, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, M.; Jonsson, P.R.; Dahlström, M.; Lundälv, T.; Burman, R.; Göransson, U.; Bohlin, L. Two Brominated Cyclic Dipeptides Released by the Coldwater Marine Sponge Geodia barrette Act in Synergy as Chemical Defense. J. Nat. Prod. 2011, 74, 449–454. [Google Scholar] [CrossRef]
- Lavey, N.P.; Coker, J.A.; Ruben, E.A.; Duerfeldt, A.S. Sclerotiamide: The First Non-Peptide-Based Natural Product Activator of Bacterial Caseinolytic Protease P. J. Nat. Prod. 2016, 79, 1193–1197. [Google Scholar] [CrossRef] [Green Version]
- Kuramochi, K.; Ohnishi, K.; Fujieda, S.; Nakajima, M.; Saitoh, Y.; Watanabe, N.; Takeuchi, T.; Nakazaki, A.; Sugawara, F.; Arai, T.; et al. Synthesis and biological activities of neoechinulin A derivatives: New aspects of structure-activity relationships for neoechinulin A. Chem. Pharm. Bull. 2008, 56, 1738–1743. [Google Scholar] [CrossRef] [Green Version]
- Ravikanth, V.; Reddy, V.L.N.; Ramesh, P.; Rao, T.P.; Diwan, P.V.; Khar, A.; Venkateswarlu, Y. An immunosuppressive tryptophan-derived alkaloid from Lepidagathis cristata. Phytochemistry 2001, 58, 1263–1266. [Google Scholar] [CrossRef]
- Usui, T.; Kondoh, M.; Cui, C.-B.; Mayumi, T.; Osada, H. Tryprostatin A, a specific and novel inhibitor of microtubule assembly. Biochem. J. 1998, 333, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Wollinsky, B.; Ludwig, L.; Hamacher, A.; Yu, X.; Kassack, M.U.; Li, S.-M. Prenylation at the indole ring leads to a significant increase of cytotoxicity of tryptophan-containing cyclic dipeptides. Bioorg. Med. Chem. Lett. 2012, 22, 3866–3869. [Google Scholar] [CrossRef]
- Jain, H.D.; Zhang, C.; Zhou, S.; Zhou, H.; Ma, J.; Liu, X.; Liao, X.; Deveau, A.M.; Dieckhaus, C.M.; Johnson, M.A.; et al. Synthesis and structure–activity relationship studies on tryprostatin A, an inhibitor of breast cancer resistance protein. Bioorg. Med. Chem. 2008, 16, 4626–4651. [Google Scholar] [CrossRef] [Green Version]
- Woehlecke, H.; Osada, H.; Herrmann, A.; Lage, H. Reversal of breast cancer resistance protein-mediated drug resistance by tryprostatin A. Int. J. Cancer 2003, 107, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.W.; Boebel, T.A.; Hartwig, J.F. Iridium-Catalyzed, Silyl-Directed Borylation of Nitrogen-Containing Heterocycles. J. Am. Chem. Soc. 2010, 132, 4068–4069. [Google Scholar] [CrossRef]
- Song, Z.; Antonchick, A.P. Iridium(iii)-catalyzed regioselective C7-sulfonamidation of indoles. Org. Biomol. Chem. 2016, 14, 4804–4808. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; Bhonoah, Y.; Frost, C.G. Beyond C2 and C3: Transition-Metal-Catalyzed C–H Functionalization of Indole. ACS Catal. 2017, 7, 5618–5627. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Maeda, R.; Iwao, M. Directed C-7 lithiation of 1-(2,2-diethylbutanoyl)indoles. Tetrahedron 1999, 55, 9151–9162. [Google Scholar] [CrossRef]
- Kona, C.N.; Nishii, Y.; Miura, M. Iridium-Catalyzed Direct C4- and C7-Selective Alkynylation of Indoles Using Sulfur-Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 9856–9860. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qiu, X.; Zhao, Y.; Mu, Y.; Shi, Z. Palladium-Catalyzed C–H Arylation of Indoles at the C7 Position. J. Am. Chem. Soc. 2016, 138, 495–498. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, P.; Zhao, Y.; Shi, Z. Regiocontrolled Direct C−H Arylation of Indoles at the C4 and C5 Positions. Angew. Chem. Int. Ed. 2017, 56, 3966–3971. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; He, Y.; Tan, L.; Ma, D. Rhodium-Catalyzed Regioselective C7-Functionalization of N -Pivaloylindoles. Angew. Chem. Int. Ed. 2016, 55, 321–325. [Google Scholar] [CrossRef]
- Lu, X.; He, S.-J.; Cheng, W.-M.; Shi, J. Transition-metal-catalyzed C H functionalization for late-stage modification of peptides and proteins. Chin. Chem. Lett. 2018, 29, 1001–1008. [Google Scholar] [CrossRef]
- Shah, T.A.; De, P.B.; Pradhan, S.; Punniyamurthy, T. Transition-metal-catalyzed site-selective C7-functionalization of indoles: Advancement and future prospects. Chem. Commun. 2019, 55, 572–587. [Google Scholar] [CrossRef]
- Demopoulos, V.J.; Nicolaou, I. Electrophilic Substitution of Indole on the Benzene Moiety: A Synthesis of 5-Acyl- and 5-Aroylindoles. Synthesis 1998, 1998, 1519–1522. [Google Scholar] [CrossRef]
- Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L.R.; Baran, P.S. Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137, 10160–10163. [Google Scholar] [CrossRef] [Green Version]
- Bandari, C.; Scull, E.M.; Bavineni, T.; Nimmo, S.L.; Gardner, E.D.; Bensen, R.C.; Burgett, A.W.; Singh, S. FgaPT2, a biocatalytic tool for alkyl-diversification of indole natural products. MedChemComm 2019, 10, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.P.; Scull, E.M.; Dimas, D.A.; Bavineni, T.; Bandari, C.; Batchev, A.L.; Gardner, E.D.; Nimmo, S.L.; Singh, S. Acceptor substrate determines donor specificity of an aromatic prenyltransferase: Expanding the biocatalytic potential of NphB. Appl. Microbiol. Biotechnol. 2020, 104, 4383–4395. [Google Scholar] [CrossRef] [Green Version]
- Bandari, C.; Scull, E.M.; Masterson, J.M.; Tran, R.H.Q.; Foster, S.B.; Nicholas, K.M.; Singh, S. Determination of Alkyl-Donor Promiscuity of Tyrosine-O -Prenyltransferase SirD from Leptosphaeria maculans. ChemBioChem 2017, 18, 2323–2327. [Google Scholar] [CrossRef] [PubMed]
- Scull, E.M.; Bandari, C.; Johnson, B.P.; Gardner, E.D.; Tonelli, M.; You, J.; Cichewicz, R.H.; Singh, S. Chemoenzymatic synthesis of daptomycin analogs active against daptomycin-resistant strains. Appl. Microbiol. Biotechnol. 2020, 104, 7853–7865. [Google Scholar] [CrossRef]
- Chen, R.; Gao, B.; Liu, X.; Ruan, F.; Zhang, Y.; Lou, Y.Z.J.; Feng, K.; Wunsch, C.; Li, S.-M.; Dai, J.; et al. Molecular insights into the enzyme promiscuity of an aromatic prenyltransferase. Nat. Chem. Biol. 2016, 13, 226–234. [Google Scholar] [CrossRef]
- Tarcz, S.; Xie, X.; Li, S.-M. Substrate and catalytic promiscuity of secondary metabolite enzymes: O-prenylation of hydroxyxanthones with different prenyl donors by a bisindolyl benzoquinone C- and N-prenyltransferase. RSC Adv. 2014, 4, 17986–17992. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Zheng, X.; Li, S.-M. Substrate Promiscuity of the Cyclic Dipeptide Prenyltransferases fromAspergillus fumigatus. J. Nat. Prod. 2009, 72, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Elshahawi, S.I.; Cao, H.; Shaaban, K.A.; Ponomareva, L.V.; Subramanian, T.; Farman, M.L.; Spielmann, H.P.; Phillips, G.N., Jr.; Thorson, J.S.; Singh, S. Structure and specificity of a permissive bacterial C-prenyltransferase. Nat. Chem. Biol. 2017, 13, 366–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, M.E. Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep. 2015, 32, 88–101. [Google Scholar] [CrossRef]
- Liebhold, M.; Li, S.-M. Regiospecific Benzylation of Tryptophan and Derivatives Catalyzed by a Fungal Dimethylallyl Transferase. Org. Lett. 2013, 15, 5834–5837. [Google Scholar] [CrossRef]
- Liebhold, M.; Xie, X.; Li, S.-M. Breaking Cyclic Dipeptide Prenyltransferase Regioselectivity by Unnatural Alkyl Donors. Org. Lett. 2013, 15, 3062–3065. [Google Scholar] [CrossRef] [PubMed]
- Liebhold, M.; Xie, X.; Li, S.M. Expansion of enzymatic Friedel-Crafts alkylation on indoles: Acceptance of unnatural beta-unsaturated allyl diphospates by dimethylallyl-tryptophan synthases. Org. Lett. 2012, 14, 4882–4885. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Liang, S.; Wu, X.-L.; Xiong, J.; Zong, M.-H.; Lou, W.-Y. Metal-organic frameworks as novel matrices for efficient enzyme immobilization: An update review. Coord. Chem. Rev. 2020, 406, 213149. [Google Scholar] [CrossRef]
- Bhushan, I.; Parshad, R.; Qazi, G.N.; Gupta, V.K. Immobilization of Lipase by Entrapment in Ca-alginate Beads. J. Bioact. Compat. Polym. 2008, 23, 552–562. [Google Scholar] [CrossRef]
- Kim, J.; Grate, J.W.; Wang, P. Nanostructures for enzyme stabilization. Chem. Eng. Sci. 2006, 61, 1017–1026. [Google Scholar] [CrossRef]
- Kumar, K.; Wang, P.; Sanchez, R.; Swartz, E.A.; Stewart, A.F.; DeVita, R.J. Development of Kinase-Selective, Harmine-Based DYRK1A Inhibitors that Induce Pancreatic Human β-Cell Proliferation. J. Med. Chem. 2018, 61, 7687–7699. [Google Scholar] [CrossRef]
- Tanaka, S.; Shiomi, S.; Ishikawa, H. Bioinspired Indole Prenylation Reactions in Water. J. Nat. Prod. 2017, 80, 2371–2378. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | K562 GI50 (µM) |
|---|---|
| TPS-A | 97 ± 21 |
| TPS-B | 78 ± 31 |
| TPS-2 | 54 ± 15 |
| TPS-24 | 50 ± 29 |
| TPS-25 | 56 ± 19 |
| TPS-46 | 37 ± 8.8 |
| TPS-52 | 77 ± 22 |
| TPS-58 | 44 ± 18 |
| TPS-61 | 89 ± 29 |
| TPS-62 | 100 ± 33 |
| TPS-65 | 53 ± 19 |
| Taxol | 0.0058 ± 0.0039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardner, E.D.; Dimas, D.A.; Finneran, M.C.; Brown, S.M.; Burgett, A.W.; Singh, S. Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts 2020, 10, 1247. https://doi.org/10.3390/catal10111247
Gardner ED, Dimas DA, Finneran MC, Brown SM, Burgett AW, Singh S. Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts. 2020; 10(11):1247. https://doi.org/10.3390/catal10111247
Chicago/Turabian StyleGardner, Eric D., Dustin A. Dimas, Matthew C. Finneran, Sara M. Brown, Anthony W. Burgett, and Shanteri Singh. 2020. "Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT" Catalysts 10, no. 11: 1247. https://doi.org/10.3390/catal10111247
APA StyleGardner, E. D., Dimas, D. A., Finneran, M. C., Brown, S. M., Burgett, A. W., & Singh, S. (2020). Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts, 10(11), 1247. https://doi.org/10.3390/catal10111247