Characterization of the Novel Ene Reductase Ppo-Er1 from Paenibacillus Polymyxa


Abstract
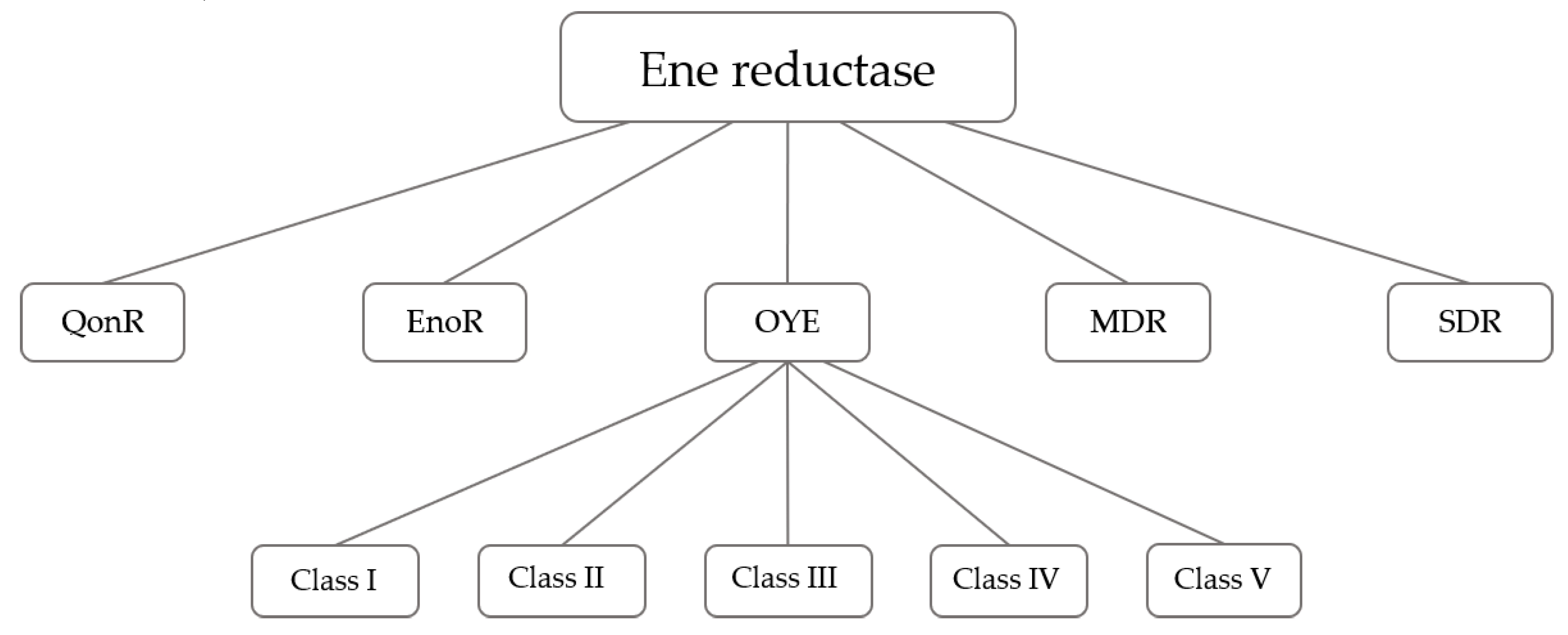
:1. Introduction
2. Results and Discussion
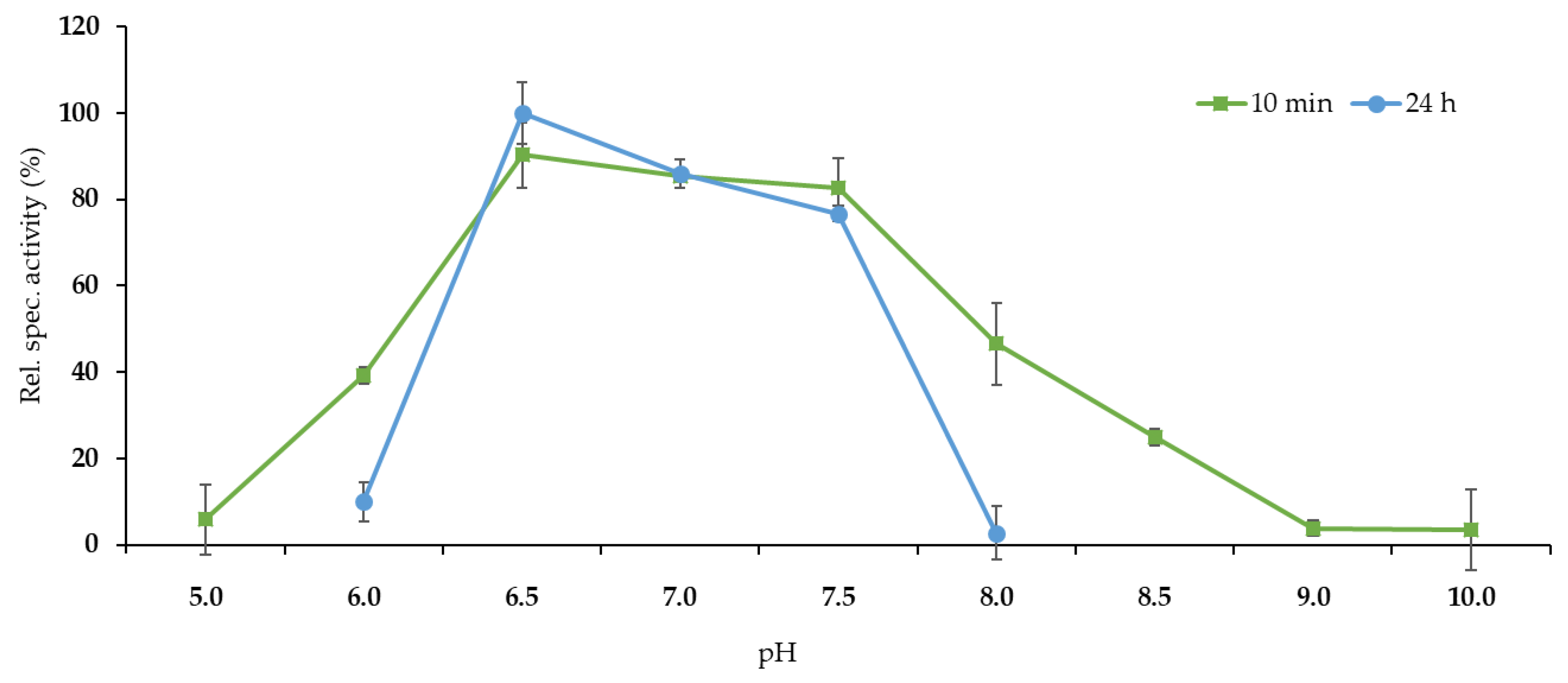
2.1. Expression and Characterization of Ppo-Er1
2.2. Substrate Scope, Determination of Michaelis–Menten Parameters, and Stereoselectivity
3. Materials and Methods
3.1. Materials
3.2. Plasmid
3.3. Bacterial Strains and Culture Conditions
3.4. Expression
3.5. Enzyme Purification
3.6. Activity Assay
3.7. Biocatalysis Reaction
3.8. GC-Analysis
3.9. Gel Filtration
3.10. Melting Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- List, B.; Yang, J.W. Chemistry. The organic approach to asymmetric catalysis. Science 2006, 313, 1584–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.N.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, M.; MacDermaid, C.; Ollis, A.A.; Chang, S.; Khan, D.; Hosford, J.; Latham, J.; Ihnken, L.A.F.; Brown, M.J.B.; Fuerst, D.; et al. Chiral synthesis of lsd1 inhibitor gsk2879552 enabled by directed evolution of an imine reductase. Nat. Catal. 2019, 2, 909–915. [Google Scholar] [CrossRef]
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.D. Biocatalysis: A pharma perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Toogood, H.S.; Scrutton, N.S. Discovery, characterisation, engineering and applications of ene reductases for industrial biocatalysis. ACS Catal. 2019, 8, 3532–3549. [Google Scholar] [CrossRef]
- Toogood, H.S.; Gardiner, J.M.; Scrutton, N.S. Biocatalytic reductions and chemical versatility of the old yellow enzyme family of flavoprotein oxidoreductases. Chemcatchem 2010, 2, 892–914. [Google Scholar] [CrossRef]
- Winkler, C.K.; Faber, K.; Hall, M. Biocatalytic reduction of activated cc-bonds and beyond: Emerging trends. Curr. Opin. Chem. Biol. 2018, 43, 97–105. [Google Scholar] [CrossRef]
- Shimoda, K.; Ito, D.I.; Izumi, S.; Hirata, T. Novel reductase participation in the syn-addition of hydrogen to the c=c bond of enones in the cultured cells of Nicotiana tabacum. J. Chem. Soc. Perkin Trans. 1996, 1, 355–358. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (nobel lecture). Angew. Chem. Int. Ed. Engl. 2002, 41, 1999–2007. [Google Scholar]
- Noyori, R. Asymmetric catalysis: Science and opportunities (nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Knaus, T.; Toogood, H.S.; Scrutton, N.S. Ene-reductases and their applications. In Green Biocatalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 473–488. [Google Scholar]
- Steinkellner, G.; Gruber, C.C.; Pavkov-Keller, T.; Binter, A.; Steiner, K.; Winkler, C.; Lyskowski, A.; Schwamberger, O.; Oberer, M.; Schwab, H.; et al. Identification of promiscuous ene-reductase activity by mining structural databases using active site constellations. Nat. Commun. 2014, 5, 4150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, K.; Buller, R. Ene-reductases in pharmaceutical chemistry. In Pharmaceutical Biocatalysis: Chemoenzymatic Synthesis of Active Pharmaceutical Ingredients; Grunwald, P., Ed.; Jenny Stanford Publishing: Singapore, 2019. [Google Scholar]
- Peters, C.; Frasson, D.; Sievers, M.; Buller, R. Novel old yellow enzyme subclasses. Chembiochem 2019, 20, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Christian, W. Ein zweites sauerstoffübertragendes ferment und sein absorptionsspektrum. Naturwissenschaften 1932, 20, 688. [Google Scholar] [CrossRef]
- Haas, E. Isolierung eines neuen gelben ferments. Biochem. Z 1938, 298, 369–390. [Google Scholar]
- Breithaupt, C.; Strassner, J.; Breitinger, U.; Huber, R.; Macheroux, P.; Schaller, A.; Clausen, T. X-ray structure of 12-oxophytodienoate reductase 1 provides structural insight into substrate binding and specificity within the family of oye. Structure 2001, 9, 419–429. [Google Scholar] [CrossRef]
- Kohli, R.M.; Massey, V. The oxidative half-reaction of old yellow enzyme: The role of tyrosine 196. J. Biol. Chem. 1998, 273, 32763–32770. [Google Scholar] [CrossRef] [Green Version]
- Fox, K.M.; Karplus, P.A. Old yellow enzyme at 2 å resolution: Overall structure, ligand binding, and comparison with related flavoproteins. Structure 1994, 2, 1089–1105. [Google Scholar] [CrossRef] [Green Version]
- Barna, T.M.; Khan, H.; Bruce, N.C.; Barsukov, I.; Scrutton, N.S.; Moody, P.C.E. Crystal structure of pentaerythritol tetranitrate reductase: “Flipped” binding geometries for steroid substrates in different redox states of the enzyme. J. Mol. Biol. 2001, 310, 433–447. [Google Scholar] [CrossRef]
- Kitzing, K.; Fitzpatrick, T.B.; Wilken, C.; Sawa, J.; Bourenkov, G.P.; Macheroux, P.; Clausen, T. The 1.3 å crystal structure of the flavoprotein yqjm reveals a novel class of old yellow enzymes. J. Biol. Chem. 2005, 280, 27904–27913. [Google Scholar] [CrossRef] [Green Version]
- Adalbjörnsson, B.V.; Toogood, H.S.; Fryszkowska, A.; Pudney, C.R.; Jowitt, T.A.; Leys, D.; Scrutton, N.S. Biocatalysis with thermostable enzymes: Structure and properties of a thermophilic ‘ene’-reductase related to old yellow enzyme. ChemBioChem 2010, 11, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Nizam, S.; Verma, S.; Borah, N.N.; Gazara, R.K.; Verma, P.K. Comprehensive genome-wide analysis reveals different classes of enigmatic old yellow enzyme in fungi. Sci. Rep. 2014, 4, 4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietruszka, J.; Schölzel, M. Ene reductase-catalysed synthesis of (r)-profen derivatives. Adv. Synth. Catal. 2012, 354, 751–756. [Google Scholar] [CrossRef]
- Li, Z.N.; Wang, Z.X.; Meng, G.; Lu, H.; Huang, Z.D.; Chen, F.E. Identification of an ene reductase from yeast Kluyveromyces marxianus and application in the asymmetric synthesis of (R)-profen esters. Asian. J. Org. Chem. 2018, 7, 763–769. [Google Scholar] [CrossRef]
- Waller, J.; Toogood, H.S.; Karuppiah, V.; Rattray, N.J.W.; Mansell, D.J.; Leys, D.; Gardiner, J.M.; Fryszkowska, A.; Ahmed, S.T.; Bandichhor, R.; et al. Structural insights into the ene-reductase synthesis of profens. Org. Biomol. Chem. 2017, 15, 4440–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryszkowska, A.; Fisher, K.; Gardiner, J.M.; Stephens, G.M. A short, chemoenzymatic route to chiral β-aryl-γ-amino acids using reductases from anaerobic bacteria. Org. Biomol. Chem. 2010, 8, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.K.; Clay, D.; Davies, S.; O’Neill, P.; McDaid, P.; Debarge, S.; Steflik, J.; Karmilowicz, M.; Wong, J.W.; Faber, K. Chemoenzymatic asymmetric synthesis of pregabalin precursors via asymmetric bioreduction of β-cyanoacrylate esters using ene-reductases. J. Org. Chem. 2013, 78, 1525–1533. [Google Scholar] [CrossRef]
- Winkler, C.K.; Clay, D.; Turrini, N.G.; Lechner, H.; Kroutil, W.; Davies, S.; Debarge, S.; O’Neill, P.; Steflik, J.; Karmilowicz, M.; et al. Nitrile as activating group in the asymmetric bioreduction of beta-cyanoacrylic acids catalyzed by ene-reductases. Adv. Synth. Catal. 2014, 356, 1878–1882. [Google Scholar] [CrossRef] [Green Version]
- Janicki, I.; Kiełbasiński, P.; Turrini, N.G.; Faber, K.; Hall, M. Asymmetric bioreduction of β-activated vinylphosphonate derivatives using ene-reductases. Adv. Synth. Catal. 2017, 359, 4190–4196. [Google Scholar] [CrossRef]
- Bertolotti, M.; Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Santangelo, S. Substrate scope evaluation of the enantioselective reduction of β-alkyl-β-arylnitroalkenes by old yellow enzymes 1–3 for organic synthesis applications. ChemCatChem 2016, 8, 577–583. [Google Scholar] [CrossRef]
- Dobrijevic, D.; Benhamou, L.; Aliev, A.E.; Méndez-Sánchez, D.; Dawson, N.; Baud, D.; Tappertzhofen, N.; Moody, T.S.; Orengo, C.A.; Hailes, H.C.; et al. Metagenomic ene-reductases for the bioreduction of sterically challenging enones. RSC Adv. 2019, 9, 36608–36614. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Yan, M.; Xu, L.; Wei, M. Identification and characterization of a novel old yellow enzyme from Bacillus subtilis str.168. J. Mol. Catal. B Enzym. 2016, 130, 18–24. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, X.; Ren, J.; Feng, J.; Zhang, T.; Wu, Q.; Zhu, D. Enzymatic hydrogenation of diverse activated alkenes. Identification of two bacillus old yellow enzymes with broad substrate profiles. J. Mol. Catal. B Enzym. 2014, 105, 118–125. [Google Scholar] [CrossRef]
- Gao, X.; Ren, J.; Wu, Q.; Zhu, D. Biochemical characterization and substrate profiling of a new nadh-dependent enoate reductase from Lactobacillus casei. Enzyme Microb. Technol. 2012, 51, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.J.; Deng, Z.; Karplus, P.A.; Massey, V. On the active site of old yellow enzyme: Role of histidine 191 and asparagine 194. J. Biol. Chem. 1998, 273, 32753–32762. [Google Scholar] [CrossRef] [Green Version]
- Spiegelhauer, O.; Dickert, F.; Mende, S.; Niks, D.; Hille, R.; Ullmann, M.; Dobbek, H. Kinetic characterization of xenobiotic reductase a from Pseudomonas putida 86. Biochemistry 2009, 48, 11412–11420. [Google Scholar] [CrossRef]
- Scholtissek, A.; Tischler, D.; Westphal, A.; van Berkel, W.; Paul, C. Old yellow enzyme-catalysed asymmetric hydrogenation: Linking family roots with improved catalysis. Catalysts 2017, 7, 130. [Google Scholar] [CrossRef]
- Hummel, W.; Gröger, H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J. Biotechnol. 2014, 191, 22–31. [Google Scholar] [CrossRef]
- Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than nature: Nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Choi, D.S.; Pesic, M.; Lee, Y.W.; Paul, C.E.; Hollmann, F.; Park, C.B. Cofactor-free, direct photoactivation of enoate reductases for the asymmetric reduction of c=c bonds. Angew. Chem. Int. Ed. 2017, 56, 8681–8685. [Google Scholar] [CrossRef] [Green Version]
- Litthauer, S.; Gargiulo, S.; van Heerden, E.; Hollmann, F.; Opperman, D.J. Heterologous expression and characterization of the ene-reductases from Deinococcus radiodurans and Ralstonia metallidurans. J. Mol. Catal. B Enzym. 2014, 99, 89–95. [Google Scholar] [CrossRef]
- Davies, M.T. A universal buffer solution for use in ultra-violet spectrophotometry. Analyst 1959, 84, 248–251. [Google Scholar] [CrossRef]
- Peters, C.; Kölzsch, R.; Kadow, M.; Skalden, L.; Rudroff, F.; Mihovilovic, M.D.; Bornscheuer, U.T. Identification, characterization, and application of three enoate reductases from Pseudomonas putida in in vitro enzyme cascade reactions. ChemCatChem 2014, 6, 1021–1027. [Google Scholar] [CrossRef]
- Pesic, M.; Fernández-Fueyo, E.; Hollmann, F. Characterization of the old yellow enzyme homolog from Bacillus subtilis (yqjm). ChemistrySelect 2017, 2, 3866–3871. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.-Y.; Pei, X.-Q.; Wu, Z.-L. Identification and characterization of a novel “thermophilic-like” old yellow enzyme from the genome of Chryseobacterium sp. Ca49. J. Mol. Catal. B Enzym. 2014, 108, 64–71. [Google Scholar] [CrossRef]
- Zheng, L.; Lin, J.; Zhang, B.; Kuang, Y.; Wei, D. Identification of a yeast old yellow enzyme for highly enantioselective reduction of citral isomers to (R)-citronellal. Bioresour. Bioprocess. 2018, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Toogood, H.; Sakuma, M.; Gardiner, J.M.; Stephens, G.M.; Scrutton, N.S. Asymmetric reduction of activated alkenes by pentaerythritol tetranitrate reductase: Specificity and control of stereochemical outcome by reaction optimisation. Adv. Synth. Catal. 2009, 351, 2976–2990. [Google Scholar] [CrossRef]
- Tischler, D.; Gadke, E.; Eggerichs, D.; Gomez Baraibar, A.; Mugge, C.; Scholtissek, A.; Paul, C.E. Asymmetric reduction of (r)-carvone through a thermostable and organic-solvent-tolerant ene-reductase. Chembiochem 2019. [Google Scholar] [CrossRef] [Green Version]
- Robescu, M.S.; Niero, M.; Hall, M.; Cendron, L.; Bergantino, E. Two new ene-reductases from photosynthetic extremophiles enlarge the panel of old yellow enzymes: Ctoye and gsoye. Appl. Microbiol. Biotechnol. 2020, 104, 2051–2066. [Google Scholar] [CrossRef]
- Riedel, A.; Mehnert, M.; Paul, C.E.; Westphal, A.H.; van Berkel, W.J.; Tischler, D. Functional characterization and stability improvement of a ‘thermophilic-like’ ene-reductase from Rhodococcus opacus 1cp. Front. Microbiol. 2015, 6, 1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.; Stueckler, C.; Ehammer, H.; Pointner, E.; Oberdorfer, G.; Gruber, K.; Hauer, B.; Stuermer, R.; Kroutil, W.; Macheroux, P.; et al. Asymmetric bioreduction of c=c bonds using enoate reductases opr1, opr3 and yqjm: Enzyme-based stereocontrol. Adv. Synth. Catal. 2008, 350, 411–418. [Google Scholar] [CrossRef]
- Josuran, R. Prot pi. Available online: https://www.protpi.ch/ (accessed on 22 January 2020).
- Forneris, F.; Orru, R.; Bonivento, D.; Chiarelli, L.R.; Mattevi, A. Thermofad, a thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J. 2009, 276, 2833–2840. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Conversion | ee | kcat/Km | Km | kcat | |
|---|---|---|---|---|---|---|
| Name | Structure | (%) | (%) | (mM−1 s−1) | (mM) | (s−1) |
| Maleimide |  | ≥99 ± 3.7 | 287.8 ± 0.12 | 0.10 ± 0.01 | 28.78 ± 0.62 | |
| trans-β-Methyl-β-nitrostyrene |  | 81 ± 1.0 | 41.4 ± 0.23 | 0.12 ± 0.03 | 4.97 ± 0.36 | |
| 2-Methyl-2-pentenal |  | ≥99 ± 7.4 | (S) 63 | 15.3 ± 0.09 | 0.41 ± 0.04 | 6.27 ± 0.11 |
| Cinnamaldehyde |  | ≥99 ± 1.5 | 14.6 ± 0.14 | 0.36 ± 0.05 | 5.27 ± 0.18 | |
| Hexenal |  | ≥99 ± 3.4 | 3.3 ± 0.10 | 2.22 ± 0.21 | 7.42 ± 0.01 | |
| Carvone |  | ≥99 ± 2.1 | (R) 98 | 0.5 ± 0.16 | 4.35 ± 0.69 | 2.20 ± 0.08 |
| Cyclohexenone |  | ≥99 ± 0.5 | 0.4 ± 0.08 | 13.42 ± 1.0 | 5.25 ± 0.1 | |
| Citral |  | 29 ± 1.4 | (S) 94 | 0.2 ± 0.93 | 1.12 ± 1.0 | 0.17 ± 0.04 |
| 2-Methyl-2-cyclohexenone |  | 76.2 ± 0.4 | (R) 92 | 0.1 ± 0.23 | 14.93 ± 3.3 | 1.30 ± 0.08 |
| Cyclopentenone |  | 59 ± 1.7 | 0.03 ± 0.17 | 57.24 ± 9.4 | 1.75 ± 0.16 | |
| 4-Phenyl-3-buten-2-one |  | ≥99 ± 1.0 | n.s. | |||
| Butylacrylate |  | 22 ± 6.5 | ||||
| Diethyl benzyldienemalonate |  | 1.2 ± 0.0 | ||||
| 3-Methyl-2-cyclohexenone |  | n.d. | ||||
| 3-Methyl-2-cyclopentenone |  | n.d. | ||||
| Etylcrotonate |  | n.d. | ||||
| Butenoic acid |  | n.d. | ||||
| Cinnamic acid |  | n.d. | ||||
| Citraconic acid |  | n.d. | ||||
| Class I | Class II | Class III | ||||||
|---|---|---|---|---|---|---|---|---|
| Substrate | PETNR | YqjM | TOYE | DrER | RmER | OYERo2 | Ppo-Er1 | YqiG |
| Cyclohexenone | 5 | 6.4 | 0.5 | 2.1 | 0.7 | 0.4 | 22 | |
| 2-Methyl-cyclohexenone | 4 | 1.0 | 0.1 | |||||
| Cyclopentenone | <0.5 | 1.9 | 0.6 | 0.03 | ||||
| Hexenal | 0.60 | 3.3 | ||||||
| Citral | 9 | 0.02 | 0.05 | 0.2 | 6.7 | |||
| 2-Methyl-2-pentenal | 61 | 0.14 | 15.3 | 18 | ||||
| Cinnamaldehyde | 8 | 14.6 | ||||||
| Carvone | 2 | 1.5 | 0.5 | 7.5 | ||||
| Maleimide | 10,800 | 287.8 | 800 | |||||
| trans-β-Methyl-β-nitrostyrene | 41.4 | |||||||
| Class I | Class II | Class III | Class IV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Substrate | OPR1 | OPR3 | PETNR | YqjM | TOYE | Ppo-Er1 | YqiG | Lla-ER | Ppo-Er3 |
| 2-Methyl-2-pentenal | (R) 47 | (S) 78 | (R) 20 | (S) 55 | (S) 63 | (S) 33 | (S) 5 | (S) 67 | |
| Carvone | (R) 95 | (R) 82 | (R) 95 | (R) 98 | (R) 89 | (R) >99.9 | (R) 91 | ||
| 2-Methyl-2-cyclohexenone | (R) 77 | (R) 62 | (R) 81 | (R) 92 | (R) 83 | (R) 11 | (R) 86 | ||
| Citral | (S) >95 | (S) >95 | (S) 95 | (S) 91 | (S) 94 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aregger, D.; Peters, C.; Buller, R.M. Characterization of the Novel Ene Reductase Ppo-Er1 from Paenibacillus Polymyxa. Catalysts 2020, 10, 254. https://doi.org/10.3390/catal10020254
Aregger D, Peters C, Buller RM. Characterization of the Novel Ene Reductase Ppo-Er1 from Paenibacillus Polymyxa. Catalysts. 2020; 10(2):254. https://doi.org/10.3390/catal10020254
Chicago/Turabian StyleAregger, David, Christin Peters, and Rebecca M. Buller. 2020. "Characterization of the Novel Ene Reductase Ppo-Er1 from Paenibacillus Polymyxa" Catalysts 10, no. 2: 254. https://doi.org/10.3390/catal10020254
APA StyleAregger, D., Peters, C., & Buller, R. M. (2020). Characterization of the Novel Ene Reductase Ppo-Er1 from Paenibacillus Polymyxa. Catalysts, 10(2), 254. https://doi.org/10.3390/catal10020254